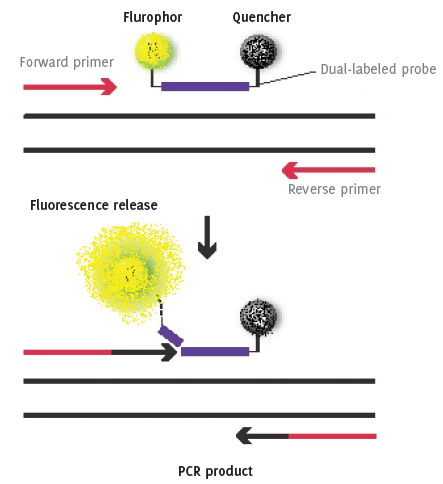
The principle of RT-PCR with dual labeled probe containing a flurophor on one end and a quencher molecule on the other.
Kordo B. A. Saeed1, Nusreen S. Ahmad2
doi: http://dx.doi.org/10.5195/ijms.2013.22
Volume 1, Number 1: 28-36
Received 15 12 2011: Accepted 19 02 2012
ABSTRACT
The polymerase chain reaction (PCR) has revolutionized the detection of DNA and RNA. Real-Time PCR (RT-PCR) is becoming the gold standard test for accurate, sensitive and fast diagnosis for a large range of infectious agents. Benefits of this procedure over conventional methods for measuring RNA include its sensitivity, high throughout and quantification. RT-PCR assays have advanced the diagnostic abilities of clinical laboratories particularly microbiology and infectious diseases. In this review we would like to briefly discuss RT-PCR in diagnostic microbiology laboratory, beginning with a general introduction to RT-PCR and its principles, setting up an RT-PCR, including multiplex systems and the avoidance and remediation of contamination issues. A segment of the review would be devoted to the application of RT-PCR in clinical practice concentrating on its role in the diagnosis and treatment of infectious diseases.
Keywords: Real-Time Polymerase Chain Reaction; Reagent Kits, Diagnostic; Molecular Diagnostic Techniques.
Polymerase chain reaction (PCR) is a molecular based technique. It derives its name from one of its key components, DNA polymerase, which is used to amplify a piece of DNA by in vitro enzyme-mediated replication. As PCR progresses, the DNA thus generated is itself used as template for replication. This in turn will set in motion a chain reaction in which the DNA template is exponentially amplified. The technique can be used to amplify single or few genomic copies of a target, which would otherwise remain undetected. A key component for this process is a pair of primers, which are composed of oligonucleotides (about 20 nucleotides in length); these are complementary to a pre-defined sequence on both the forward and reverse strands of DNA. The same primers can be used again and again, to make other copies of the target sequence. A revolutionary step in the process was the discovery of a thermo-stable DNA polymerase, Taq polymerase, from Thermophilus aquaticus a bacterium that can withstand extreme temperatures, allowing the reaction to continue without the addition new polymerase in every round of the process. For conventional PCR, 35 cycles of amplification are usually carried out, and the product can be visualized in the form of “bands” on agarose gel. This is a qualitative approach, as often the amount of product is not related to the amount of input DNA.
Real time-PCR (RT-PCR), a technique that follows the general basics of conventional PCR however it allows the measurement of PCR products in real time as the reaction progresses. Reverse transcriptase PCR analysis of RNA, which can also mistakenly be abbreviated as “RT-PCR”, is an analysis to measure RNA. Here RNA is copied to complementary DNA (cDNA) by reverse transcriptase. This product the is amplified by PCR allowing the formation of a second strand of DNA, which can be further amplified and eventually analyzed conventionally or in real time. In this review the abbreviation RT-PCR represents real-time PCR.
There are many different commercial platforms available for nucleic acid extraction. The choice of extraction platform will depend on the needs of the laboratory in terms of the quantity of samples received. Low, medium and high throughput automated extraction platforms can be widely found from most companies, which can be used to process up to 96 samples in one extraction run. In most cases the chemistry for RNA (as RNA is less stable, reagents are added to stabilize the extracted nucleic acid) and DNA extraction will differ but the extraction process will remain the same.
The extraction process usually begins by lysing the cells of the target organism using enzymes such as proteinase K. Once lysed, the nucleic acid can then be bound to a membrane or to glass beads which are then washed several times to remove any possible inhibitors that may be present in the original sample and the remaining enzymes used for lysis. Once washing is complete, the nucleic acid can then be eluted off in to a clean collection tube. This extract is then ready for PCR. At this stage the eluate can be stored at −20°C for a short period of time if PCR is not planned immediately. Manual extraction processes also follow the same format of lysing, binding, washing and eluting.1–3
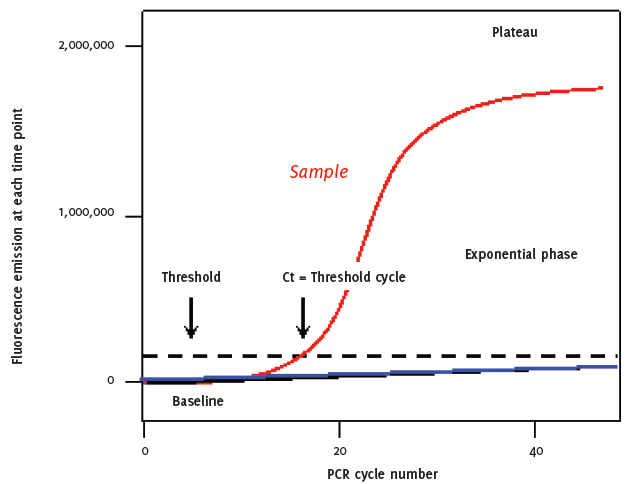
As with conventional PCR, in RT-PCR, double strand DNA (ds-DNA) is denatured at approximately 95°C, this high temperature breaks the hydrogen bonds that bind one side of the helix to the other allowing the two strands separate. The sample is cooled to between 50 to 60°C to allow annealing of primers that are complementary to a specific site on each strand. The temperature is raised to 72ºC and the addition of the heat-stable Taq polymerase extends the DNA from the primers generating four cDNA strands. A major improvement in this system is the addition of a dual labeled probe, which contains a flurophor on one end and a quencher molecule on the other. The shape of the un-bound probe allows the flurophor and quencher molecules to sit in close proximity to each other preventing the premature release of fluorescence. After each cycle, the level of fluorescence released is measured when bound to the ds-DNA i.e. the PCR product. The progress of PCR amplification can be continuously monitored in real time by acquiring fluorescence signals in each amplification reaction cycle. Thus, after 30 to 40 cycles, quantitative information of the PCR process is obtained by plotting the intensity of the fluorescence signal versus cycle number (Figure 1 and 2). Automated assays are relatively easy to perform and they are not only valuable in reducing chances of contamination, but also enable reproducibility and rapid processing of multiple samples
Figure 1.The principle of RT-PCR with dual labeled probe containing a flurophor on one end and a quencher molecule on the other.
Graphic representation of RT-PCR quantitative plot after 40 cycles, quantitative information of the PCR process is obtained by plotting the intensity of the fluorescence signal versus cycle number.
Duplex, triplex and multiplex RT-PCRs are reactions in which two, three or more fluorogenic probes are used simultaneously for the discrimination of multiple amplicons in a single tube. These reactions are possible because probes labeled with fluorescent dyes with different excitation and emission spectra allowing each target to me measured independently of the others. The main advantages of multiplexing over single-target analysis are the ability to provide internal controls, lower reagent costs and preservation of precious samples. The introduction of combinatorial fluorescence energy transfer tags is important in boosting the development of multiplex real-time PCR.4
There are two quantifications types from RT-PCR,5–7 one of which is an absolute quantification that requires an input standard curve with series diluted template (five- or tenfold serial dilution). This curve is used as a reference standard for extrapolating quantitative information for target nucleic acid. Using the known copy level of the standard reagent, the software of the PCR instrument generates a standard curve in a plot that relates fluorescence (measure of amplified product) and the cycle number in which the nucleic acid target is detected. This method determines the absolute amount of a target (expressed as copy number or concentration), which is one of the most accurate standards for gene expression analysis.
Relative quantification, also known as the comparative threshold Method determines the ratio between the amount of target and an endogenous reference gene, usually a suitable housekeeping gene, that are not expected to change under the experimental conditions serve as a convenient internal standard. This normalized value can then be used to compare, for example, differential gene expression in different samples. The target and the reference genes are amplified from the same sample, either separately or in the same reaction (duplex RT-PCR). The normalized value is determined for each sample and can be used to compare, for example, differential expression of a gene in different tissues. Because the absolute quantity of the internal standard is not known, only relative changes can be determined by this method. If a housekeeping gene cannot be found whose amplification efficiency is similar to the target, then the standard curve method should be used.
The key to real-time PCR is the ability to monitor amplification as it occurs. This is accomplished by specific chemistries and instrumentation. Generally, chemistries consist of special fluorescent probes in the PCR. Several types of probes exist, including DNA-binding dyes like SYBR green I which intercalate in to the ds-DNA as it is formed, hydro-lysis probes (5_-nuclease probes), hybridization probes, molecular beacons, sunrise and scorpion primers, and peptide nucleic acid (PNA) light-up probes. Each type of probe has its own unique characteristics, but the strategy is they must link a change in fluorescence to amplification of DNA.
One class of RT-PCR chemistry uses different fluorescent dyes incorporated in short oligonucleotide probes specific for the amplified target. Another class consists of dyes that bind ds-DNA and become fluorescent; the most commonly used of these is SYBR Green I.8 SYBR Green I fluorescence is enormously increased upon binding to the minor groove ds-DNA. As it is an intercalating fluorescent dye, during the extension phase, more and more SYBR Green I will bind to the PCR product, resulting in an exponential rise on fluorescence. Consequently, during each subsequent PCR cycle more fluorescence signal will be detected. The technique is inexpensive and generic, as it requires the same detection reagent for each template to be tested. But detection with dyes like SYBR Green I are less specific than probe-based detection methods. In addition, SYBR Green I cannot be used in multiplexed assays. The benefit of using this type of assay in a non clinical setting is to investigate the possibility of contamination during the validation process. This method can be used to visualize the melting temperatures of various targets that may have been inadvertently introduced in to the PCR mix as they are likely to have a different melting temperature to the actual target in question. This process can replace the laborious task of pouring and running conventional gels to look for contamination. It is worth noting that SYBR Green is not commonly used in routine clinical practice.
Hydrolysis probe technique: Addressing the specificity problem, these are sequence-specific and conjugated with a quencher fluorochrome, which absorbs the fluorescence of the reporter fluorochrome on condition that the probe is intact. However, upon amplification of the target sequence, the hydrolysis probe is displaced and subsequently hydrolyzed by the Taq polymerase. This results in the separation of the reporter and quencher fluorochrome and consequently the fluorescence of the reporter fluorochrome becomes detectable. Throughout the PCR cycles this fluorescence will further increase because of the progressive and exponential buildup of free reporter fluorochromes.1 This allows results to be visualized in real-time which in a clinical setting can be important.
TaqMan probes depend on the 5’-nuclease activity of the DNA polymerase used for PCR to hydrolyze an oligonucleo-tide that is hybridized to the target amplicon. The probe has a fluorescent reporter dye attached to its 5´ end and a quencher dye at its 3´ terminus. If the target sequence is present, the fluorogenic probe anneals downstream from one of the primer sites and is cleaved by the 5´ nuclease activity of the Taq polymerase enzyme during the extension phase of the PCR. Whilst the probe is intact, fluorescence resonance energy transfer (FRET) occurs and the fluorescence emission of the reporter dye is absorbed by the quenching dye. During PCR, when the polymerase replicates a template on which a TaqMan probe is bound, the 5’-nuclease activity of the polymerase cleaves the probe. This separates the reporter and quencher dyes, thereby increasing the fluorescence from the former, which has a linear relationship with the amount of probe cleavage. Well-designed TaqMan probes require very little optimization, and can be used for multiplex assays. There are several other variations on the reporter-quencher theme, including molecular Beacons, sunrise primers, and scorpion primers. It is possible to convert a monoplex assay in to a sequence of multiplex assays allowing more than one target to be identified in one single reaction. This is important in a clinical setting especially when dealing with samples such as cerebrospinal fluid which are difficult to collect and often come in small volumes. Each extraction may only elute as little as 50µl so being the ability to test multiple targets whilst using the minimal amount of eluted nucleic acid is an advantage.
Hybridization probes technique: In this technique one probe is labelled with a donor fluorochrome at the 3’ end and a second–adjacent-probe is labeled with an acceptor fluorochrome. When the two fluorochromes are in close vicinity (1–5 nucleotides apart), the emitted light of the donor fluorochrome will excite the acceptor fluorochrome FRET. This results in the emission of fluorescence, which subsequently can be detected during the annealing phase and first part of the extension phase of the PCR reaction. After each subsequent PCR cycle more hybridization probes can anneal, resulting in higher fluorescence signals. This method is widely been used for detection of minimal residual disease after treatment and viral load quantification.1
The chemistries and instrumentation of RT-PCR are closely connected. The instrumentations that are required for RTPCR technique consists of a thermal cycler for amplification, a light source for excitation of fluorescent probes, a camera for recording, and a computer to control instruments and record data. Because fluorescent chemistries require both a specific input of energy for excitation and a detection of a particular emission wavelength, the instrumentation must be able to do both simultaneously and at the desired wavelengths. Scientists can be faced with a daunting task when selecting platforms as machines are available from several manufacturers, with difference in sample capacity (e.g. 96-well standard format, some require specialized glass capillary tubes), methods of excitation, some use lamp (broad-spectrum emission devices), light-emitting diode (LED), or laser (narrow spectrum), and overall sensitivity. There are also platform-specific differences in how the software processes data. Emission energies must also be detected, by cameras or other types of photodetectors, at the appropriate wavelengths in order to collect appropriate data. Narrow wavelength filters or channels are generally employed to allow only the desired wavelength(s) to pass to the photodetector to be measured. The detector and the capability of the instrument for distinguishing different wavelength signals can influence the level of assay and allows for multiplexing.
Efficient performance of quantitative PCR requires rapid, precise, thermal control. Hence, the significance of the thermocycler in RT-PCR is in its ability to maintain a consistent temperature among all sample wells, as any differences in temperature could lead to erroneous PCR amplification efficiencies. The temperature uniformity has a direct impact on the ability to discriminate between different PCR products by performing melting point analysis. Additionally, the resolution with which instruments are able to control temperature is a major factor, which affects their performance when performing high resolution melting analysis. Thermal non-uniformity during temperature cycling contributes to variability in PCR and can affect the RT-PCR accuracy. This can be solved by using a heating block, heated air, or a combination of the two. It is important to ask respective manufacturers to provide up to date information on the platform you chose to purchase for your facility. To ensure consistent analysis and reporting of results, suitable computer hardware, data-acquisition and analysis software are essential to complete the process.9
PCR cannot take place without a number of specific reagents, that include;
The PCR is commonly carried out in a reaction volume of 20–150 µl in small reaction tubes (0.2–0.5 ml volumes) in a thermal cycler. The thermal cycler heats and cools the reaction tubes to achieve the temperatures required at each step of the reaction (see below). Many modern thermal cyclers make use of the Peltier effect which permits both heating and cooling of the block holding the PCR tubes simply by reversing the electric current. Thin-walled reaction tubes permit favorable thermal conductivity to allow for rapid thermal equilibration. Most thermal cyclers have heated lids to prevent condensation at the top of the reaction tube. The efficacy of RT-PCR is determined by its specificity, low background fluorescence, steep fluorescence increase, high amplification efficiency, and high level plateau.10–13
Contamination remains an issue for laboratories performing detection of microbes using PCR. RT-PCR processing can be classified into two major groups, the pre-PCR stage (including preparation of both the sample and PCR reagents) and post-PCR (PCR execution and analysis). Compliance with guidelines to prevent contamination is essential for successfully operating a PCR laboratory on a long-term basis. Guidelines must be part of a network of protocols or standard operating procedures focused on maintaining a contamination-free zone in the laboratory. Contamination sources vary, e.g. one of the most frequent causes of PCR product contamination is the generation of aerosols of PCR amplicons that is associated with the post-PCR analysis. Other sources include previous procedures and amplified molecules (“amplicons”). Purification of plasmid clones, repeated isolation of template nucleic acids should not be ignored as potential sources; DNA templates are typically more troublesome as contaminants because they are more stable than RNA targets. There are a number of approaches to control of PCR contamination, and the stringency that is required in a laboratory is invariably determined by the assay being performed. A practical approach to control contamination is to segregate activities and limit the performance of PCR activities to its own area. The crucial steps in preventing, eliminating or controlling contamination include separation of pre- and post-PCR procedures, utilization of physical barrier approaches e.g. maintaining a unidirectional flow of work in the laboratory, the use of barrier pipette tips to prevent aerosols generation, or use of chemical means e.g. use of ultraviolet photolinking, use of aliquoted PCR reagents, incorporation of numerous positive and negative or blank PCRs (H2O substituted for template), and use of Uracil-DNA-glycosylase (UDG) an enzyme effective at destroying PCR amplicons when vigorously used for sample preparation. Use of consistent, careful technique by the operator coupled with monitoring of PCR controls will ensure a proactive approach to PCR contamination.
This can often be caused by poor quality oligonucleotides, a badly extracted sample (resulting in poor quality template), insufficient sample (where the nucleic acid may be too diluted to be detected) or by expired or poor quality reagents that are not suitable for the RT-PCR platform. It is necessary to include both positive and negative controls as well as an extracted internal positive control in order to determine both the extraction efficiency and the presence of inhibitors in the sample and the efficiency of the PCR.
Can be observed when gene expression is low and can be easily resolved by increasing concentration of the target template. Primers that have been designed to be less than 24 base pairs long and which are not rich in “GC” content, ensuring that there are no more than three “G’s” together and also preventing the use of “G” oligonucleotides at the 5’ end often will help to prevent the formation of primer dimers (Figure 3).
Figure 3.Primer dimer can be observed in RT-PCR when gene expression is low and can be easily resolved by increasing concentration of the target template.
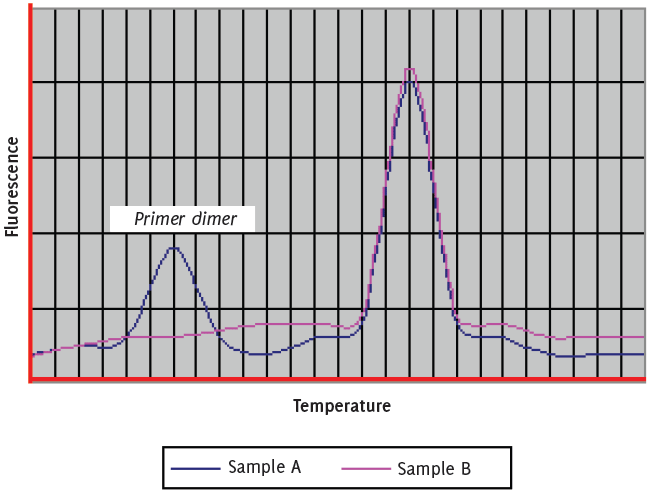
The presence of spurious bands in both melt curve analysis and agarose gel electrophoresis is most likely to occur due to poor assay optimisation. In this case it is advisable to investigate further both the cycling conditions (non specific binding can occur if the annealing/extension temperature in a “two step” PCR is far lower than the combined melting temperature of both the primers and probe) and chemistry of the assay. It is also necessary to “check” the oligonucleo-tide sequences to ensure that those selected are common only to the target organism.
RT-PCR has the ability to quantify nucleic acids over a wide dynamic range (7–8 log units). This is coupled to high technical sensitivity, allowing the detection of less than five copies of a target sequence. With appropriate internal standards and calculations, mean variation coefficients are 1–2%, allowing reproducibility. In addition, all real-time platforms are relatively quick, with some affording high-throughput automation. RT-PCR is performed in a closed reaction vessel that requires no post-PCR manipulations, thereby minimizing the chances for cross contamination in the laboratory.
Like other diagnostics there are several limitations to RTPCR methods. RT-PCR is susceptible to inhibition by compounds present in certain biological samples. Because of the necessary use of RNA in an extra enzymatic step, errors can occur. RNA itself is extremely labile compared with DNA, and therefore isolation must be carefully performed to ensure both the integrity of the RNA itself and the removal of contaminating nucleases, genomic DNA, and reverse transcriptase or PCR inhibitors. This can be a problem with any sample source, but clinical samples are of special concern because inconsistencies in sample size, collection, storage, and transport can lead to a variable quality of RNA templates. Conversion of RNA to cDNA during the RT reaction is also subject to variability because multiple reverse transcriptase enzymes with different characteristics exist, and different classes of oligonucleotides can be used to prime RT.
Probably the largest present limitation of RT-PCR, however, is not inherent in the technology but rather resides in human error: improper assay development, incorrect data analysis, or unwarranted conclusions. RT-PCR primer sets must be designed and validated by stringent criteria to ensure specificity and accuracy of the results. For microbiology, false positives or negatives must be considered when designing an assay to detect pathogens. Amplification and melting curves must be visually inspected while independent calculations based on these curves should be double-checked for accuracy. Of course, conclusions based on data derived from RT-PCR are best utilized when the biological context is well understood.
RT-PCR is widely used across many clinical disciplines for example in clinical oncology RT-PCR has been used for the detection and quantification of chromosomal translocations, to monitor minimal residual disease or to show graft-versus lymphoma effects.14–16 Other applications include predictive genetic testing and the identification of relevant single nucleotide polymorphisms. Another major area for applications of real-time reverse transcriptase RT-PCR as-says is the quantification of gene expression.
In this section we would like to concentrate on the application of RT-PCR in clinical microbiology where highly conserved regions of the target organism genome tend to be chosen for assay development to avoid detection failure especially in agents with high mutation rates. The concern is with the emerging infectious agents e.g. severe acute respiratory syndrome coronavirus (SARS-CoV), where comparative genome investigations should be performed before a reliable assay can be developed.1,17,18
Results of RT-PCR assays for detection bacteria can inform the clinician of the infection status of the patient more rapidly than culture methods. This is of particular importance as in the current era of rising antimicrobial resistance; it allows more specific and timely administration of appropriate and directed antimicrobial therapy. This will not only increase the treatment efficiency but also reduce the time of hospitalization, and avoid the inappropriate use of antibiotics with unwanted effects.
PCR has long been the preferred method for diagnosis of bacteria that are difficult to culture, and numerous RT-PCR assays have already been developed to replace conventional PCR methods for bacteria such as Legionella pneumophila (which can take up to fourteen days for cultures to grow), Chlamydiophilia (which can only be cultured in active tissue cell lines) and Mycobacterium spp (can take weeks to grow). The method has now become an important tool in the rapid detection bacterial pathogens implicated as biological weapons. An excellent example is the successful application of RT-PCR for screening for the presence of Bacillus anthracis spores and for differentiation based on virulence encoding plasmids and chromosomal markers.
Some RT-PCR assays may also be useful in deciding on the use of antimicrobial agents. The rapid turnaround time of RT-PCR can detect and monitor the antibiotic resistance of clinical isolates of bacteria such as, enterobacteriacae (e.g. Extended spectrum Beta lactamse producers ESBL or Metallo Beta lactamse producers), Staphylococcus aureus, S. epidermidis, Helicobacter pylori, Enterococcus faecalis, and Enterococcus faecium (Glycopeptide resistant enterococci or GRE or VRE). For example, rapid RT-PCR analysis detecting rifampin resistance in M. tuberculosis complex, and a RTPCR was applied successfully to screen for MRSA from nasal swabs and joint fluids.19,20 RT-PCR is becoming one of the main tests proposed for multi-step algorithms in testing for Clostridium difficile genes in suspected cases with negative immuno-assays.21 Another important area is the ability to extract E. coli O157:H7 DNA, directly from stool specimens, which is not only important for patient management but also for prompt epidemiological investigations. A specific PCR for detecting Neisseria meningitides, Haemophilus influenzae, and Streptococcus pneumoniae has provided more rapid results than culture, especially where antibiotics are administered before samples were obtained.22
RT-PCR can complement histopathological and serological analysis, e.g. detection of Bartonella henselae, a difficult to culture organism, in patient samples with suspected Cat Scratch Disease. As is the case for other fastidious organisms, PCR offers an attractive alternative for detecting Bordetella pertussis and B. parapertussis in clinical specimens. PCR can be used to detect Tropheryma whipplei genes from a variety of human specimens, allowing appropriate treatment with antibiotics for appropriate duration. Further more, the laboratory diagnosis and screening for Borrelia burgdorferi (Lyme borreliosis) is currently based on serology which has some drawbacks e.g. false negatives mainly during the early stages of the infection and false positive results especially in some patients with autoimmune disease. PCR on various tissues and body fluids, including the CSF, may have the potential to be helpful to tackle these issues, however, details of patient sample requirements and rapid sample preparation techniques remain to be worked out.23–26
Since its discovery, RT-PCR has had a major impact on clinical virology. This is mainly due to the less genetic complexity of these organisms, the lack of rigid cell membranes makes it easier to extract nucleic acids from viruses and traditionally the effort of conventional culture is great for viruses.
Qualitative Viral Assays have been applied in the diagnosis of Herpes simplex virus (HSV), data has shown increased detection rates by RT-PCR for up to 300% over cell culture methods for diagnosis of HSV infections, with faster turnaround times at equivalent cost.27 Varicella-zoster virus (VZV): RT-PCR techniques permit highly sensitive same-day detection of VZV in clinical specimens, e.g. in testing CSF samples for the diagnosis of VZV meningitis or encephalitis. Enteroviruses: Collectively, these viruses are associated with diverse clinical manifestations ranging from mild febrile illness to aseptic meningitis, encephalitis, myocarditis, and poliomyelitis.
Early detection of these agents in clinical samples have shown valuable in reducing medical costs incurred by patients by reducing hospitalization and hospital stays and the use of unnecessary antibiotics and antiviral agents.28 Other applications are in the diagnosis of respiratory Viruses, including Adenovirus, Influenza virus types A and B, Parainfluenza virus, Respiratory syncytial virus (RSV), SARS-CoV and Human Metapneumovirus. Rapid laboratory diagnosis of influenza is critical for infection control, especially in hospital and nursing home settings intervention with effective antiviral treatment if provided to the patient in the early stages of this viral disease.
Conventional techniques e.g. cell cultures, are hazardous in some cases e.g. SARS-CoV, because of the risk of laboratory-acquired infections with this agent and bio-safety level 3 laboratory facilities are required for culturing the virus Commercial RT-PCR reagents are critical not only for the rapid recovery and identification of SARS-CoV, but also for reducing transmission to healthcare workers and laboratory personnel. RT-PCR using throat or nasopharyngeal samples also played a major role in early diagnosis during the 2009–10 “pandemic” of H1N1 or “swine flu”. These tests have improved the capabilities of hospital and public health laboratories for diagnosing viral respiratory tract infections and emerging infections and assisted public health agencies in identifying outbreaks and pandemics.29–31
Quantitative Viral Assays have been used to assess the relationship between the viral load (copy level) and the prediction of progression of infection and development of clinical disease. For example in Cytomegalovirus (CMV) using serial samples from transplant cases to monitor CMV DNA/ml, by RT-PCR, have been used to detect evolving symptomatic infection or for initiating preemptive antiviral therapy. Similarly quantification of Epstein-Barr Virus (EBV) DNA provides the potential for the designation of viral load levels generally associated with healthy or subclinical carriers of EBV (reactivated infection) compared with those levels of virus that produce disease states such as post transplant lymphoproliferative disorder in transplant patients.32,33
Human Immunodeficiency Virus (HIV): HIV-1 and HIV-2 RNA levels in the plasma of infected individuals can be determined reliably by quantitative rapid RT-PCR assays. These assays will help clinicians in early identification of patients who are virologically responding to highly-active antiretroviral therapy (HAART) and achieve HIV viral load copies below 50 copies/mL.34,35 There are a number of other assays e.g. for Hepatitis B virus which is important for monitoring disease progression and for assessing the response to anti-viral therapy36 and also quantitative
Hepatitis B virus PCR is been used as a guide employers e.g. healthcare workers who are infected with Hepatitis B virus in the UK and whose viral load rose above 103 genome-equivalents/ml should are restricted from performing exposure prone procedures for as long as their viral load remained above that level.37
Over the past 20 years, dramatic advancements have occurred in the molecular approach to the investigation of parasites and parasitic diseases. RT-PCR methodology has been efficiently used to detect, differentiate, and diagnose parasites that infect humans and animals. Among parasitic infections, RT-PCR has been applied most vigorously in the diagnosis and detection of drug resistance genes of Plasmodium spp - causative agents of malaria. Specific RT-PCRs have been developed to diagnose Toxoplasma gondii, Trypanosoma cruzi, Babesia spp., Leishmania spp., Cryptosporidium parvum, Entamoeba, and Giardia spp. The traditional culture method, animal inoculation, and serological detection are not only time consuming but also lack of sensitivities. RT-PCR vastly improved speed and sensitivity of diagnosis of such parasites with impact on clinical management and epidemiological investigations.38–45
RT-PCR assays can also be successfully applied to the diagnosis of diseases caused by Aspergillus fumigatus and A. flavus. The rationale behind this effort is that timely detection of Aspergillus spp. may decrease the extreme morbidity and mortality associated with invasive aspergillosis by initiating antifungal therapy in a timely manner, particularly in immunocompromised individuals. RT-PCR can also detect A. fumigatus mutations that confer high-level resistance to antifungals.
The use of RT-PCR has also been of great use in identification of Cryptococcal disease which has become a major fungal infection in patients infected with HIV and AIDS patients. In addition PCR has been used in the early identification of Pneumocystis jirovecii the causative organism on Pneumocystis pneumonia (PCP).
High sensitivity of this method makes the assay appealing in the identification of this organism in immunocompromised patients which can help clinicians confirming their clinical suspicion on the basis of risk factors, Chest-XRay findings and pulse oximetry which ultimately leads to early initiation of appropriate treatment that in turn can improve outcome and shorten the patient’s hospital stay.46–48
Numerous technological advances have been made using PCR since its discovery in the mid-1980’s. RT-PCR is one of the enabling technologies of the genomic era and has become the method of choice or the gold standard for the detection of nucleic acid. Prior to the development RT-PCR, quantitative PCR was very labor intensive. Real-time technology has significantly extended the use and scope of RTPCR assays, with the potential for quantification of nucleic acid targets a particular advantage. RT-PCR offers streamlined assay development, reproducible results, and a large dynamic range. The focus on nucleic acid quantification, together with the introduction of advanced uncomplicated instrumentation and chemistries, has facilitated the migration of this technology from research laboratories into individual diagnostic laboratories. Despite some of the difficulties that may still need to be worked out, the foundation has been set for the use of quantitative RT-PCR in routine diagnostic laboratories. This is reflected in the increasingly significant role it plays in clinical diagnosis in infection and infectious diseases, in particular when used to determine the viral load and disease progression. The technology can generate large amounts of data within a relatively short time. The accuracy of the obtained data is largely depended on several factors including; the choice of chemistries, primers and probes, and instruments. Appropriate application, quality control and standardization are also important matters and must be considered, it is vital to take time to evaluate each stage of the development protocol, starting with laboratory setup, sample acquisition and template preparation, and the RT-PCR steps. Only if all of these stages are appropriately validated is it possible that molecular diagnosticians can reliably interpret data to clinical colleagues to have the best positive impact on patients’ outcome.
We would like to thank Editorial Staff of the International Journal of Medical Students, for giving us this opportunity to write this review.
The Authors have no funding, financial relationships or conflicts of interest to disclose.
1. Espy MJ, Uhl JR, Sloan LM, et al. Real-Time PCR in Clinical Microbiology: Applications for Routine Laboratory Testing. Clin Microbiol Rev 2006; 19(1):165–256.
2. Swift GH, Peyton MJ, MacDonald RJ. Assessment of RNA quality by semiquantitative RT-PCR of multiple regions of a long ubiquitous mRNA. Biotechniques 2000; 28(3):524–31.
3. Mannhalter C, Koizar D, Mitterbauer G. Evaluation of RNA isolation methods and reference genes for RT-PCR analyses of rare target RNA. Clin Chem Lab Med 2000; 38(2):171–7.
4. Saeed K, Ahmad N, Pallett A, Guiver M, Marsh P. Specific staphylococcal polymerase chain reaction can be a complementary tool for identifying causative organisms and guiding antibiotic management in orthopaedic infections. Curr Orthop Pract 2010; 21(6):628–31.
5. Orlando C, Pinzani P, Pazzagli M. Developments in quantitative PCR. Clin Chem Lab Med 1998; 36(5):255–69.
6. Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 2000; 25(2):169–93.
7. Pfaffl MW, Hageleit M. Validities of mRNA quantification using recombinant RNA and recombinant DNA external calibration curves in real-time RT-PCR. Biotechn Lett 2001; 23:275–282.
8. Abdul-Careem MF, Hunter BD, Nagy E, et al. Development of a real-time PCR assay using SYBR Green chemistry for monitoring Marek’s disease virus genome load in feather tips. J Virol Methods 2006; 133(1):34–40.
9. Comprehensive list of vendors. Accessed at various time points. Available from: <http://www.appliedbiosystems.com>, <http://www.bioneer.co.kr>, <http://discover.bio-rad.com>, <http://www.eppendorf.com/mastercycler/index.html>, <http://www.stratagene.com>, <http://www.techne.com/CatMol/quantica.htm>, <http://www.appliedbiosystems.com>, <http://www.roche-applied-science.com>, <http://www.biotrove.com>, <http://www.corbettresearch.com)>, <http://www.cepheid.com>, <http://www.biobank.co.kr/pcr/insyte.shtml>, <http://www.fluidigm.com>, <http://www.alphahelix.com/pages/superconvector.html>,<http://www.dna-technology.com/catalog/ob_dt322_en.shtml>,<http://www.bioer.com.cn/en/shengming_66pcr.htm>,.
10. Premier Biosoft International. Accessed on 24.10.2011. Available from: <http://www.premierbiosoft.com/tech_notes/real_time_PCR.html> and Primer Bank: PCR Primers for Gene Expression Detection and Quantification. available from: <http://pga.mgh.harvard.edu/primerbank/>.
11. Protocol online. Accessed on 24.10.2011. Available from: <http://www.protocol-online.org/prot/Molecular_Biology/PCR/Real-Time_PCR/>.
12. The Gene Quantification page: Summarises all technical aspects involved in quantitative gene expression analysis using RT-PCR. It presents a lot of new and innovative PCR applications, chemistries, methods, algorithms, QC strategies, cyclers, kits, dyes, analysis methods, events, and services involved. Accessed on 24.10.2011. Available from: <http://www.gene-quantification.info/>.
13. Dennis Lo YM. Setting up a PCR laboratory. Methods mol med 1998; 16:11–7.
14. Corradini P, Astolfi M, Cherasco C, et al. Molecular monitoring of minimal residual disease in follicular and mantle cell non-Hodgkin’s lymphomas treated with high-dose chemotherapy and peripheral blood progenitor cell autografting. Blood. 1997;89:724–731.
15. van Dongen J, Macintyre EA, Gabert JA, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 concerted action investigation of minimal residual disease in acute leukemia. Leukemia 1999;13(12):1901–28.
16. Mandigers CM, Meijerink JP, Raemaekers JM, Schattenberg AV, Mensink EJ. Graft-versus-lymphoma effect of donor leucocyte infusion shown by real-time quantitative PCR analysis of t(14;18). Lancet 1998; 352(9139):1522–3.
17. Bustin S A and Mueller R. Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin Sci (Lond) 2005;109(4):365–79
18. Kaltenboeck B, Wang C. Advances in real-time PCR: application to clinical laboratory diagnostics. Adv Clin Chem 2005; 40: 219–59.
19. Helb D, Jones M, Story E, et al. Rapid Detection of Mycobacterium tuberculosis and Rifampin Resistance by Use of On-Demand, Near-Patient Technology. J Clin Microbiol 2010;48:229–37.
20. Jonas D, Speck M, Daschner FD, et al. Rapid PCR-based identification of methicillin-resistant Staphylococcus aureus from screening swabs. J Clin Microbiol. 2002; 40:1821–1823.
21. Department of Health/UK. Advisory Committee on Antimicrobial Resistance and Healthcare Associated Infection (ARHAI). UPDATED GUIDANCE ON THE DIAGNOSIS AND REPORTING OF CLOSTRIDIUM DIFFICILE. First published March 2012 Published to DH website, in electronic PDF format only. Available at: www.dh.gov.uk/publications.
22. Corless CE, Guiver M, Borrow R, Edwards-Jones V, Fox AJ, Kaczmarski EB. Simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae in suspected cases of meningitis and septicemia using real-time PCR. J Clin Microbiol 2001; 39(4):1553–8.
23. Zeaiter Z, Fournier PE, Greub G, Raoult D. Diagnosis of Bartonella endocarditis by a real-time nested PCR assay using serum. J. Clin. Microbiol 2003; 41:919–925.
24. Chan EL, Antonishyn N, McDonald R, et al. The use of TaqMan PCR assay for detection of Bordetella pertussis infection from clinical specimens. Arch Pathol Lab Med 2002; 126:173–176.
25. Fenollar F, Fournier PE, Raoult D, Gérolami R, Lepidi H, Poyart C. Quantitative detection of Tropheryma whipplei DNA by realtime PCR. J Clin Microbiol 2002;40(3):1119–20.
26. Pietila J, He Q, Oksi J, Viljanen MK. Rapid differentiation of Borrelia garinii from Borrelia afzelii and Borrelia burgdorferi sensu stricto by LightCycler fluorescence melting curve analysis of a PCR product of the recA gene. J. Clin. Microbiol 2000; 38:2756–2759.
27. Saeed K, Pelosi E. Comparison between turnaround time and cost of herpes simplex virus testing by cell culture and polymerase chain reaction from genital swabs. Int J STD AIDS. 2010;21(4):298–9.
28. Nigrovic LE, Chiang VW. Cost analysis of enteroviral polymerase chain reaction in infants with fever and cerebrospinal fluid pleocytosis. Arch Pediatr Adolesc Med. 2000;154(8):817–21.
29. Mahony J, Chong S, Merante F, et al. Development of a respiratory virus panel test for detection of twenty human respiratory viruses by use of multiplex PCR and a fluid microbead-based assay. J Clin Microbiol 2007; 45(9):2965–70.
30. Shu B, Wu KH, Emery S, Villanueva J, et al. Design and performance of the CDC real-time reverse transcriptase PCR swine flu panel for detection of 2009 A (H1N1) pandemic influenza virus. J Clin Microbiol. 2011;49(7):2614–9.
31. Junhui Zhai, Thomas Briese, Erhei Dai, et al. Real-Time Polymerase Chain Reaction for Detecting SARS Coronavirus, Beijing, 2003. Emerging Infectious Disease Journal. 2004;10(2):300–303.
32. Mengelle C, Pasquier C, Rostaing L. Quantitation of human cytomegaloviruse in recepiant of solid organ transplant by real-time Quantitative PCR. J Med Virol 2003;69(2):225–31.
33. Gulley ML. Molecular diagnosis of Epstein-Barr virus-related diseases. J Mol Diagn 2001;3(1):1–10.
34. Havlir DV, Bassett R, Levitan D, et al. Prevalence and predictive value of intermittent viremia with combination hiv therapy. JAMA 2001; 286(2):171–9.
35. Smith CJ, Staszewski S, Sabin CA, et al. Use of viral load measured after 4 weeks of highly active antiretroviral therapy to predict virologic outcome at 24 weeks for HIV-1-positive individuals. J Acquir Immune Defic Syndr 2004;37(1):1155–9.
36. Garson JA, Grant PR, Ayliffe U, Ferns RB, Tedder RS. Real-time PCR quantitation of hepatitis B virus DNA using automated sample preparation and murine cytomegalovirus internal control. J Virol Methods 2005;126(1–2):207–13.
37. Department of health/Uk. Guidance. Hepatitis B infected healthcare workers and antiviral therapy. 16 March 2007. Available at: http://www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_073164.
38. Farrugia C, Cabaret O, Botterel F, et al. Cytochrome b Gene Quantitative PCR for Diagnosing Plasmodium falciparum Infection in Travelers. J Clin Microbiol 2011;49(6):2191–5.
39. Berry A, Benoit-Vical F, Fabre R, et al. PCR-based methods to the diagnosis of imported malaria. Parasite. 2008;15:484–488.
40. Mei-Hui L, Tse-Ching C, Tseng-tong K, et al. Real-Time PCR for Quantitative Detection ofToxoplasma gondii. J. Clin. Microbiol. November 2000;38(11): 4121–5.
41. Piron M, Fisa R, Casamitjana N, López-Chejade P, et al. Development of a real-time PCR assay for Trypanosoma cruzi detection in blood samples. Acta Trop. 2007;103(3):195–200.
42. Teal AE, Habura A, Ennis J, Keithly JS, Madison-Antenucci S. A new real-time PCR assay for improved detection of the parasite Babesia microti. J Clin Microbiol. 2012;50(3):903–8.
43. Wortmann G, Hochberg L, Houng HH, Sweeney C, et al. Rapid identification of Leishmania complexes by a real-time PCR assay. Am J Trop Med Hyg. 2005;73(6):999–1004.
44. Verweij JJ, Blange RA, Templeton K, et al. Simultaneous detection of Entamoeba histolytica, Giardia lamblia, and Cryptosporidium parvum in fecal samples by using multiplex real-time PCR. J Clin Microbiol 2004; 42:1220–23.
45. Roy S, Kabir M, Mondal D, Ali IK, Petri WA Jr, Haque R. Real-time-PCR assay for diagnosis of Entamoeba histolytica infection. J Clin Microbiol 2005;43(5):2168–72.
46. Costa C, Vidaud D, Olivi M, Bart-Delabesse E, Vidaud M, Bretagne S. Development of two real-time quantitative TaqMan PCR assays to detect circulating Aspergillus fumigatus DNA in serum. J Microbiol Methods 2001; 44:263–9.
47. Veron V, Simon S, Blanchet D, Aznar C. Real-time polymerase chain reaction detection of Cryptococcus neoformans and Cryptococcus gattii in human samples. Diagn Microbiol Infect Dis 2009;65(1):69–72.
48. Botterel F, Cabaret O, Foulet F, Cordonnier C, Costa JM, Bretagne S. Clinical significance of quantifying Pneumocystis jirovecii DNA by using real-time PCR in bronchoalveolar lavage fluid from immunocompromised patients. J Clin Microbiol 2012 ;50(2):227–31.
Kordo B. A. Saeed, 1 Royal Hampshire County Hospital, Winchester, UK.
Nusreen S. Ahmad, 2 Health Protection Agency and Southampton University Hospitals Foundation Trust, Southampton, UK.
Correspondence Dr. Kordo Saeed. Address: Royal Hampshire county Hospital, Romsey Road, SO22 5DG UK. E. mail: kordosaeed@nhs.net
Cite as: Saeed KBA, Ahmad SN. Real-Time Polymerase Chain Reaction: Applications in Diagnostic Microbiology. Int J Med Students 2013;1(1):28-36.
Copyright © 2013 Kordo B. A. Saeed, Nusreen S. Ahmad
International Journal of Medical Students, VOLUME 1, NUMBER 1, April 2013