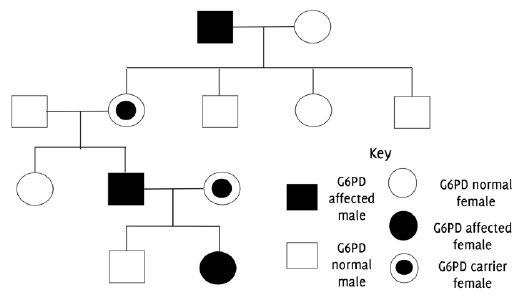
Pedigree chart indicating X-linked inheritance pattern of G6PD deficiency.
Nidhruv Ravikumar1, Graeme Greenfield2
doi: http://dx.doi.org/10.5195/ijms.2020.637
Volume 8, Number 3: 281-287
Received 01 07 2020: Rev-request 01 09 2020: Rev-request 23 11 2020: Rev-recd 19 09 2020: Rev-recd 24 11 2020: Accepted 25 11 2020
ABSTRACT
Deficiency of the glucose-6-phosphate dehydrogenase (G6PD) enzyme is a common X-linked disorder that affects people globally. It was first identified in the 1950s as a disorder that primarily affects the red blood cells, causing a myriad of symptoms including acute hemolytic anemia, neonatal jaundice and chronic nonspherocytic hemolytic anemia. This deficiency has been extensively studied and, especially within the last 5 years, there have been improvements in the diagnosis and management. Various methods of diagnosis exist; however, recent research focuses on the use of biosensors for a more accurate and less time-consuming diagnosis. Guidelines suggest controlling symptomology, as no specific treatment currently exists. A common complication of the disease is neonatal jaundice, and research on phototherapy has proved to show some effect in managing this condition. In the last year, protein-protein interactions have been used as targets to enhance enzyme stability and activity. AG1 is a small molecule activator that has demonstrated effectiveness in treating G6PD deficiency in models. This review summarizes existing literature and potential areas of research on glucose-6-phosphate dehydrogenase deficiency including clinical characteristics, diagnosis, and management.
Keywords: Glucosephosphate Dehydrogenase deficiency; Hemolytic anemia; Neonatal jaundice; Erythrocytes (Source: MeSH-NLM).
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is one of the commonest forms of enzymatic disorders affecting humans worldwide. Four-hundred million people across the globe are estimated to be affected, with the highest prevalence being in those of African, Asian and Mediterranean descent.1,2 G6PD is an enzyme in the pentose phosphate pathway (PPP) that catalyzes the first step.3 PPP plays a role in reductive biosynthesis, cell proliferation and processing of antioxidants. The products of this pathway are important in nucleotide synthesis, lipogenesis, cholesterol synthesis and glutathione reduction. Disruption of the PPP will lead to a generation of free radicals that cause oxidative damage to red blood cells (RBCs) and, in turn, hemolysis (breakdown of RBCs).4
G6PD deficiency was first identified in the 1950s a small number of American soldiers who developed a hemolytic anemia upon exposure to anti-malarial drugs. In later years, the genetic cause of the disease was identified and was attributed to mutations in the g6pdx gene located on the X chromosome.5 Since the gene is located on the X chromosome, it is inherited in a sex-linked manner (Figure 1). Males have only one copy of the X chromosome; hence, those who inherit the mutation are considered hemizygous and will develop the condition. However, females can be either homozygous (two pairs of mutant alleles) or heterozygous (only one mutant allele). Homozygous females will be G6PD deficient, while heterozygous females will be carriers. Carriers are not usually affected by the condition and rarely present with symptoms.3 The majority of the population with the deficiency, mainly males, do not develop symptoms unless they are exposed to certain oxidative stressors (i.e., fava beans), oxidative medications (i.e, antimalarial agents) and infections.3,5 The geographical distribution of the condition is similar to sickle cell anemia and thalassemia, and corresponds to the global distribution of malaria. This has led to the hypothesis that G6PD deficiency offers some protection against the parasite Plasmodium falciparum, a cause of malaria.6 This review thus seeks to evaluate existing literature regarding the pathophysiology, diagnosis, and management of G6PD deficiency, and provide insight into potential areas for further research.
Figure 1Pedigree chart indicating X-linked inheritance pattern of G6PD deficiency.
A comprehensive literature search was performed through PubMed resources to identify the articles which discussed the prevalence, pathophysiology, diagnosis and management of G6PD deficiency. Keywords used includeed ‘Glucose-6-phosphate dehydrogenase deficiency' or ‘G6PD deficiency', ‘Favism' and ‘Congenital hemolytic anemia'. The search terms were used as keywords and in combination as MeSH terms to maximize the output from literature findings. In conjunction to this, a manual search of reviews and other relevant studies was conducted. A staged literature search was done, whereby a separate literature search was performed for each section within this article and all the relevant studies were identified and summarized separately. The relevant articles are cited and referenced within each section separately. No limits were placed on publication time of the article.
It is estimated by The National Organization for Rare Disorders that there are four hundred million people who are living with G6PD deficiency. An estimate suggests that approximately 8% of the world's population carry the alleles responsible for the development of G6PD deficiency, ranging from a maximum of 35% in regions of Asia to 0.1% in Japan and certain parts of Europe, which makes it a significant public health problem.7 It is observed to have higher prevalence in Asia, the Middle East, Latin America and the Mediterranean (Figure 2).1 Since the mode of inheritance is X-linked, males seem to be more affected, compared to females; however, the most common genotype is a heterozygous female.3 In the past it was noted that there was an increased prevalence of the disease in high-risk countries which often correlated to the demographics observed with malaria. However, in recent times due to significant globalization and the accompanying migration of individuals, there has been a considerable change in population demographics. Hence, in the majority of countries, there exists pockets of population groups that are affected by the disorder, causing an increase in prevalence of this disorder in countries where it would be considered rare in the past.9 In the United States, for instance, people of African-American descent are observed to be predominantly affected by the disorder. In recent years, the frequency of the affected population is as high as about 10-14%.1
Figure 2Regional prevalence estimates of G6PD deficiency (Data collected from: Nkhoma E, Poole C, Vannappagari V, Hall S, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: A systematic review and meta-analysis. Blood Cells Mol Dis. May-Jun 2009;42(3):267-78).8

The high prevalence of the condition increases its probability of being associated with other pathological conditions, particularly those affecting RBCs (sickle cell hemoglobinopathy and thalassemia).5
Pharmacogenetics is the process in which the genetic determinants of an individual's response to drugs are measured. In fact, G6PD deficiency was discovered using this technique. In the 1950s, primaquine, an antimalarial drug, was produced in large-scale when the US military was deployed to areas endemic to malaria. The army physicians observed that, apart from the predicted side effects of gastrointestinal complaints, some soldiers developed jaundice and anemia. This toxic side effect was then termed as primaquine sensitivity syndrome. In the years following, Brewer et al.,10 tested the effects of primaquine on human volunteers, which provided valuable insight into the mechanism of the condition, despite its lack of ethical compliance. The group conducted enzymatic analysis and concluded that there was a marked reduction in the levels of the G6PD enzyme in the RBCs of primaquine-sensitive patients, thus coining the term G6PD deficiency. Further genetic analysis of the condition revealed that the gene coding for the enzyme is located on the X chromosome, prompting an X-linked inheritance pattern.11 Prior to this discovery, favism was a well-known condition that caused similar symptoms in certain populations upon consumption of fava beans. It was later concluded that the underlying biochemical effect was the same as that caused by primaquine.
In a G6PD-deficient person, no abnormalities are observed in the steady state. This comes down to the role of G6PD in the RBCs.11 It is an enzyme present in the cytosol that is involved in the PPP by controlling the entry of glucose-6-phosphate (G6P) into the pathway. G6PD oxidizes G6P to 6-phosphogluconolactone and, in the process, reduces NADP+ to NADPH (Figure 3).12 Hence, its main role is to provide reductive potential by the production of NADPH.11 This reaction is the rate determining step of the pathway, and therefore, decreased activity of G6PD causes reduced levels of NADPH.12 NADPH is a key reductive agent that maintains glutathione in its reduced state (Figure 3). Glutathione in its reduced form is important in scavenging potentially harmful reactive oxygen species (ROS) and confers vital protection to RBCs.13 Excess ROS have detrimental effects on the cell function –causing an increase in erythrocyte membrane fragility leading to hemolysis, release of hemoglobin into the plasma and systemic nitric oxide scavenging leading to vasoconstriction.13 This is caused by its interference in reactions involving proteins, nucleotides and lipids.12
Figure 3Oxidative stage of Pentose Phosphate Pathway (PPP) indicating deficiency of Glucose-6-phosphate dehydrogenase leads to decreased NADPH levels. In the glutathione reduction pathway, decreased NADPH causes accumulation of GSSG (oxidized glutathione), that prevents the conversion of H2O2 to H2O leading to its accumulation (Reactive oxygen species).

Although G6PD deficiency affects every cell of the body, its primary effects are blood related, as RBCs do not have an alternate source of NADPH. Non-RBC cells, on the other hand, have other non-specific enzymes (hexose-6-phosphate dehydrogenase) that work in a similar way to G6PD to generate NADPH. In the steady state, the levels of NADPH in the RBCs are adequate. However, exposure to oxidative stressors poses a challenge to G6PD-deficient RBCs, due to the generation of excess ROS.11 This leads to the lysis of RBCs, causing hemolytic anemia.13 As with other RBC enzymes, the activity of G6PD reduces as the cell ages. This is not endangering in G6PD-normal red cells; however, in G6PD-deficient cells, the older RBCs have lower levels of the enzyme, as compared to the younger cells. This then correlates to the reduced RBCs life span and acute haemolysis.7 A classification system can thus be formulated based on the variation in enzyme activity levels.
The level of disease severity varies depending on residual enzyme activity and substrate binding, which are influenced by the type of mutation.13 The World Health Organization (WHO)7 categorizes the disease into the following 5 classes:
A− and Med are common alleles that cause moderate to mild severe deficiency and they belong to class II and III. Patients affected by the A+ and B variants fall under class IV and class V. Most people affected by the deficiency fall in Class II and III, i.e., moderate deficiency and have fewer problems.7,10 The symptoms observed and prevalence of each class is summarized in Table 1.
Table 1.Classification of G6PD Deficiency.
| Class | Variant | Enzyme Activity | Symptoms | Prevalence |
|---|---|---|---|---|
| I | Mediterranean type | < 1% | Chronic non-spherocytic anemia | Uncommon across populations |
| II | A−/Med | 1% - 10% | Intermittent acute hemolysis | More common in Asian and Mediterranean populations |
| III | A−/Med | 10% - 60% | Intermittent acute hemolysis | US black males |
| IV | A+/B | 60% - 150% | No clinical symptoms | Rare |
| V | A+/B | > 150% | No clinical symptoms | Rare |
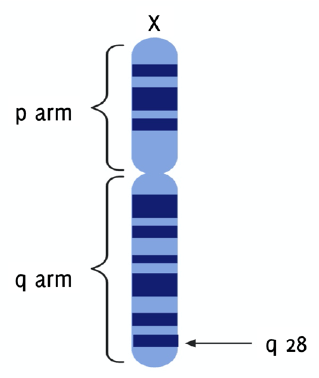
The gene that codes for G6PD, g6pdx, is situated proximal to the telomeric end of the distal arm (q arm) at band 28 of the X chromosome in humans (Xq28) (Figure 4).7,14-15 This gene has 13 exons and 12 introns, spans 18.5 Kilobases, and has a GC rich promoter region that encodes a product of 1545 bp.2,16,17 It has a high rate of heterogeneity, and over 300 variants have been found. Only 217 precise mutations, however, are currently known.1,11,14,18 Majority of the mutations observed are point mutations that occur in the coding DNA, introns and 5' and 3' untranslated regions.14 S188F (Mediterranean mutation) in the Arab population, C131G and G487A in Bangladesh, and A376G in North America, Africa, Yemen and Saudi Arabia are the commonest mutations discovered in G6PD-deficient patients.2,19-21 These variants can be either polymorphisms, with milder clinical signs, or sporadic variants. The frequency of variant polymorphs is higher than sporadic variants, 50% to 1% respectively. Nevertheless, the frequencies of sporadic variants have reached polymorphic frequencies, due to some protection offered against malaria caused by Plasmodium falciparum.
Figure 4g6pdx Gene Located on the Telomeric End of the q Arm at Band 28 of the X Chromosome (Xq28).
All mutations cause a decrease in enzyme stability.14 Active G6PD enzyme occurs as a homodimer or homotetramer. The majority of the mutations in the gene cause a change in the amino acid sequence of the enzyme, thus decreasing stability. The postulated mechanism for this decrease in stability is disturbance in protein folding. This affects particularly the dimer interface, causing a disturbance in effective dimerization. Moreover, it was also observed that a structural NADP molecule is an essential moiety that maintains the structural integrity of the enzyme. Mutations that alter the binding of this structural NADP molecule also contribute to the decrease in stability of the enzyme.5
Males are predominantly affected, as the gene is present on the X chromosome. They can be either hemizygous normal or hemizygous deficient. In contrast, females can be homozygous normal, homozygous deficient or heterozygous. A mosaic pattern of the wild type and the deficient variant is observed in heterozygous females, caused by X chromosome inactivation (i.e., lyonization). This leads to the presence of differing levels of normal and deficient RBCs. Due to the random nature of this phenomenon, the proportions of deficient variant vary, resulting in a spectrum of symptomology.16 If the proportion of deficient RBCs is greater than 50% in heterozygous females, they become more susceptible to hemolysis, but with less severity when compared to homozygous deficient females and hemizygous deficient males.
The majority of the population with a polymorphic G6PD gene does not manifest any clinical symptoms. In fact, with sporadic variants, the chance of developing acute hemolysis is 1 in 106, which is particularly infrequent.5 For the greater part of their lifespan, people with the disease are asymptomatic; however, symptoms manifest when the body is subjected to oxidative stress. The most common stressors include medications and infection, while diabetic ketoacidosis (DKA) is also a precipitator in certain variants.2 Although the mechanism of action of DKA is not known, the increased risk of infection that accompanies DKA is postulated to link DKA with specific variants of G6PD deficiency.3
Typically, G6PD deficiency presents as acute hemolytic anemia, chronic nonspherocytic hemolytic anemia, neonatal jaundice and favism.5 The most common presentation is acute hemolytic anemia, with symptoms of anemia, fatigue, back or abdominal pain, jaundice and hemoglobinuria occurring promptly after exposure to an oxidative stress. Hemolytic anemia is characterized by premature lysis of RBCs.3 Due to the polymorphic character of G6PD deficiency, a number of clinical presentations are observed in different individuals. Different mutations in the gene lead to variable severity and the associated symptoms tend to vary in a similar fashion. For instance, people with the Mediterranean mutation often fall under Class I and develop very severe clinical signs such as chronic nonspherocytic hemolytic anemia (CNSHA).5
An important complication of G6PD-deficiency is neonatal jaundice, which can occur in neonates 2 to 3 days after birth with varying severity. Neonates with the G6PD mutation also inherit a variant allele for uridine-diphosphate-glucuronosyltransferase 1 (UGT1A1).16 This mutation is responsible for Gilbert syndrome, where the conjugation of bile and glucuronic acid is impaired, leading to a condition where the levels of bile in the body are very high (i.e., hyperbilirubinemia).5,16 This increases the risk of babies developing jaundice. If hyperbilirubinemia is not treated effectively, bilirubin encephalopathy (kernicterus) can result and potentially leave the neonate with permanent neurological damage or may lead to death.16
Favism is another precipitating disorder in individuals with G6PD deficiency. The compounds innately present in fava beans, divicine, and isouramil, are strong oxidizing agents, thereby placing the cells under oxidative stress. In addition, neonates exposed to fava beans, through breast milk or passively, can soon develop neonatal jaundice.16
In males with the rare G6PD sporadic variants with severe deficient function, chronic nonspherocytic hemolytic anemia (CNHSA) is a recognized complication. This occurs when there is a lack of NADPH in the steady state for maintaining the levels of oxidizing agents. The excess build-up of oxidizing agents in the cell damages the RBC membrane, resulting in reduced RBC lifespan and chronic haemolysis.5
A variety of different disease processes often clinically manifest in a similar fashion to that of G6PD deficiency, characterized primarily by hemolytic anemia. Therefore, it is important to consider the following diseases in the differential diagnosis:1
In order to prevent the occurrence of hemolytic episodes, the presence of G6PD-deficiency should always be considered in populations of high-risk background, including people of African, Asian or Mediterranean descent. It is important to note that screening for the deficiency needs to be performed in such individuals prior to commencement of any treatment that may induce an oxidative stress. Although people carrying the variant allele for the disorder are usually asymptomatic, the influence of a particular oxidative stressor may prove to be fatal. Hence the deficiency should be considered, especially in those with a family history of recurrent jaundice, splenomegaly, cholelithiasis, or anemia. Individuals who experience hemolytic anemia after exposure to a known oxidative stressor, those with characteristic blood film findings and neonates who have jaundice should also be considered for diagnostic testing of G6PD deficiency.3
Different methods of diagnosis can be used. Clinically, quantitative measurement of the enzyme function provides the required information to classify G6PD-deficiency.22 The most widely used tests are the fluorescent spot test (FST) and the quantitative spectrophotometric assay, both of which rely on the principle of conversion of NADP+ to NADPH.5 Identification of the genetic mutations through molecular analysis can confirm the exact underlying genetic defect.3
The FST is the first line diagnostic tool that is used in patients suspected with G6PD deficiency.14,22 It is a quick and inexpensive test that is often used in regions where the deficiency and malaria are more prevalent, ensuring that the patient will not experience hemolytic anemia upon exposure to antimalarial drugs.5 Although inexpensive, FST requires the use of special equipment like UV light, a water bath or heat block, and a dark room.3 To combat the difficulty of accessing special equipment, researchers recently developed new techniques that do not require such apparatus.5 The formazan-based spot test is the most commonly used method, which can detect less than 50% of normal G6PD activity.3 The formazan method uses a substrate of 3-(4,5-Dimethylazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) and a hydrogen carrier, phenazine methosulphate (PMS). A formazan substrate is formed from these substrates under the influence of the NADPH that G6PD produces. New studies have demonstrated that the water-insoluble formazan is formed by MTT, therefore making the quantitative analysis of G6PD activity difficult. Tantular and Kawamoto23 have now discovered a new formazan substrate, 2-(2-methoxy-4-nitrphenyl)-3-(4-nitrophenyl)-5-(2,4-disulphophenyl)-2H tetrazolium monosodium salt (WST-8), that does not react with hemoglobin and is water soluble. They have proved that the WST-8 formazan-based spot test is a less time consuming and more accurate method of detecting G6PD deficiency.23
At the laboratory level, the spectrophotometric assay is the gold standard in diagnosing the deficiency. It uses the hemolysate of blood (resulting product from the lysis of RBCs) to detect the total enzymatic activity. Enzyme activity is measured as a change in the absorbance at 340 nm at a given temperature, for a given hemoglobin concentration (unit of enzyme per grams of hemoglobin; IU/gHb) or, in rare cases, for a given number of RBCs (unit of enzyme per RBC; U/RBC).24 Normal activity is considered to be 7 to 10 IU/gHb.3 This technique is dependent on the concentration of hemoglobin in the blood. A disruption in the levels of hemoglobin can therefore lead to abnormal results. In individuals with anemia or iron deficiency, the G6PD activity will be falsely elevated.24
Due to the disparities in enzyme activity through spectrophotometric assay measurement, recent studies have focused on flow cytometry as an alternative option.24 Earlier models of flow cytometry exploited the capacity of G6PD to reduce oxidized methaemoglobin (MetHb) to normal hemoglobin. Newer models are based on the capacity of G6PD to reduce MTT insoluble tetrazolium salts.24 The principle behind flow cytometry corresponds to the light scattering ability of the moving particles and fluorescence emission. Fluorescence emission is the primary property that is measured in determining the levels of G6PD.25 Tetrazolium salts are stained with a fluorescent dye.24 The levels of the enzyme are directly proportional to the amount of fluorescent probe bound to the salt.25 This is a laborious process and requires several hours for processing the sample and staining it. Nowadays, however, a simpler and quicker flow cytometric assay using the same principle has been developed.24
A study conducted by Bancone et al.,26 has shown that the flow cytometric assay and spectrophotometric assay are equally reliable for the diagnosis of G6PD deficiency in patients with normal hemoglobin and without anemia. However, they proved that flow cytometric assay is the tool of choice in anemic patients and those with other haemoglobinopathies, as it is not influenced by hemoglobin concentration.
The spectrophotometric assay for G6PD-deficiency testing is fairly expensive and requires skilled operators, an equipped laboratory and a reliable source of material. Ironically, these are available in hospitals only and are inaccessible in tropical countries, where the disease is highly prevalent.26 Point-of-care rapid diagnostic tests (POC RDT) is now considered a relatively inexpensive diagnostic tool. It is a quantitative test, and the most commonly used ones are BinaxNOW and CareStart.3 These devices are biosensors and have been developed in recent years to provide a more accurate diagnosis.26 The activity of G6PD can be obtained quantitatively in 4 minutes by these handheld devices to devise a suitable treatment plan.27 In a study comparing the diagnostic power of CareStart and spectrophotometric assay, it was demonstrated that CareStart has a higher diagnostic power, with appreciable sensitivity and specificity.26 Even though technicians easily operated CareStart, certain changes in the model were required to make it more user-friendly.26 With improvements, POC RDTs shall be used more often in detecting G6PD deficiency.
The timing of quantitative assay is crucial, as this can influence the results of the test drastically. During active periods of hemolysis, the bone marrow response falsely elevates the levels of the enzyme, due to consequent reticulocytosis. Therefore, measurement at the steady state is important.
The aforementioned techniques are useful and accurate in the detection of homozygous males and females, yet they fail to diagnose accurately in heterozygous females with intermediate enzyme activity (due to lyonization).5 In these cases, an accurate diagnosis can be made by molecular analysis methods. Molecular analysis involves sequencing the g6pdx gene to detect known mutations that cause the disease.3 Different methods have been employed for the simple detection of the common mutations. Classical molecular techniques include restriction enzyme analysis (PCR-RFLP), amplification refractory mutation system (ARMS), gradient gel electrophoresis (DGGE), denaturing high-performance liquid chromatography (DHPLC), probe melting curve and reverse dot blot assay.28
Once the diagnosis of G6PD deficiency is made, the most important next step is to discontinue the use of the offending oxidative stressor, most commonly a medication. Most patients recover spontaneously but should undergo careful monitoring. Bone marrow function in these patients tends to be normal; hence, they respond by increasing the production of RBCs. This causes a reduction in age of the circulating RBCs, making them less susceptible to hemolysis. This process plays a role in the self-limiting nature of the deficiency, thus limiting the need for management.
However, there are circumstances that require management. Firstly, if the anemia caused by the stressor is very severe, then a blood transfusion can be lifesaving. Anemia can be severe if the patient had pre-existing anemia, or if the stressor caused a rapid decline in the level of hemoglobin. There are no consensus guidelines on the use of transfusion in acute hemolytic anemia caused by G6PD. One proposal put forth by Luzzatto et al.,11 suggests that if the level of hemoglobin is less than 70 g/L, consideration should be given to a blood transfusion. If hemoglobin is less than 90 g/L, and there are signs of significant hemolysis (hemoglobinuria), then immediate blood transfusion should again be considered. If there are no signs of hemoglobinuria, blood transfusion is not necessary, rather the patient should be monitored closely for 48 hours.11 Consideration of co-morbidities, hemodynamic stability and the patients previous hemolytic experience may help guide the decision to initiate transfusion.
Acute kidney injury is a possible complication that occurs with severe acute hemolytic anemia. This is due to a situation, such as in the case of hypovolemic shock, and not because of damage caused by hemoglobinuria. Renal function usually recovers with time; however, the patient needs to be put on hemodialysis in some cases.11
Moreover, in children with severe anemia, administration of oxygen and fluids is critical. They might also need to have healthy blood cells transfused.29 There are no specific treatments for the management of G6PD deficiency, rather avoiding the oxidative stressor is the recommended management plan.
Patient education about avoiding the triggers and recognition of signs and symptoms is critical in the management of the deficiency and to prevent further exacerbations. This is particularly important in patients with severe deficiency and lower enzymatic activity. Advice regarding diet and lifestyle modifications are usually given to the patient to prevent complications.9
Neonatal jaundice is a common condition that occurs in children that are affected with G6PD deficiency. In order to manage this condition, phototherapy is a technique that is often used.30 The probability pf developing severe hyperbilirubinemia is a deciding factor for the initiation of phototherapy. An increased risk of brain damage due to hyperbilirubinemia is present in G6PD deficiency, thus potentiating the need for phototherapy in the management of neonatal jaundice.31 Phototherapy is a technique that uses visible light in the treatment of hyperbilirubinemia in neonates. This intervention involves reducing serum bilirubin by transforming it into water soluble isomers.32 It is traditionally provided by fluorescent lamps, with radiance of 296 µw/cm2 in the 400-480 nm range and 250 µw/cm2 in the 425-475 nm range, directly on the infant's skin. Phototherapy has been proved to be effective in prophylaxis along with management of neonatal jaundice.30 It may be used often, however, in certain cases where the adverse reactions caused are detrimental. It can cause acute side effects, including interference in the mother-child relationship, thermal and hydro-electrolytic imbalance, skin lesions and bronze baby syndrome, in addition to late side effects.31,33
The nature of the treatment is such that the neonate has to be separated from the mother for a period of time, thus limiting skin-skin contact and breastfeeding.31 Moreover, there exists an increased risk of vulnerable child syndrome, wherein parental reactions to a life-threatening illness can cause long-term psychological effects on the children.
A change in the thermal environment occurs within the neonate upon initiation of phototherapy. Subsequently, insensible water loss, hypothermia or hyperthermia and dehydration are potential side effects that may occur. It has also been noted that there exists reduced absorption of water, sodium and potassium. However, this is often transient, and resolves upon discontinuation of the treatment. Dehydration is seen as a serious complication, and thus in order to prevent dehydration during the course of the treatment, it is recommended to increase water supply by 10 to 15 ml/kg/day.31
Calcium levels are also closely monitored in neonates on phototherapy, due to an increase in urinary calcium excretion. Moreover, light affects calcium homeostasis by inhibiting melatonin secretion from the pineal gland leading to hypocalcaemia. The levels of calcium tend to return to normal within 24 hours after ending the treatment.34
Macules, papules and a maculopapular rash may often be caused as a consequence of phototherapy. In cases of haemolytic disease, such as G6PD-deficiency, an increased risk of purpuric eruption exists, due to simultaneous transfusion of blood and intravenous immunoglobulins.31 Phototherapy also poses an increased occurrence of common and atypical nevi as was demonstrated by Csoma et al.35
There were multiple in vitro studies that were conducted on the risk of phototherapy causing subclinical carcinogenic risk hypothesising that a wavelength of 400-450 nm would pose a problem in phototherapy.36-39 This was thought as it induces photosensitization of riboflavin which is an endogenous substance. However, this was proved wrong by a group of researchers who demonstrated the shielding effect of the Soret band of haemoglobin at wavelengths of 400-500 nm.33 There were 2 studies conducted by the same group of researchers to demonstrate the risk of developing cancer with phototherapy. Both studies demonstrated that relative risk of developing cancer with phototherapy is statistically insignificant.40,41 With the control of confounding variables, the association of cancer and phototherapy can be eliminated, but due to the possibility of partial causality, the use of phototherapy should be controlled.41
Phototherapy is also known to cause bronze baby syndrome (BBS) in certain neonates. This is an uncommon alteration in the colour of the skin (dyschromia) that occurs as a consequence of phototherapy.42 Kopelman et al., reported the first case of BBS in 1972.43 Increased accumulation of photoisomers of bilirubin, metabolic precursors or degradation products, or copper-porphyrin conjugates is the proposed cause of BBS. However, the true mechanism remains unknown. Despite the occurrence of BBS, it is still indicated to continue phototherapy in neonatal jaundice due to the innately toxic effects of excess conjugated bilirubin.42
A group of researchers, in 2019, looked at devising therapeutic measures for G6PD deficiency.44 They postulate that the use of small molecule activators of the enzyme can promote the activity of the enzyme in deficient patients. The primary principle behind this is protein-protein interactions (PPIs). PPIs are the reason for the immense signalling networks that are present for normal cellular functions. These molecular interactions together are termed as the interactome, and intervening in these is prime for modifying disease pathogenesis. Apart from signalling events, the interactome comprises of interactions between the quaternary structure of proteins to modulate enzyme stability, activity and function.44 Hence the use of small molecules that target this aspect of the interactome is essential in enhancing enzymatic activity of G6PD.
The primary cause of pathogenicity in G6PD deficient individuals are mutations that affect the homodimer interface of the enzyme. This is because only the homodimer and homtetrameric forms of the enzyme are active and stable. Raub et al.,44 identified AG1, a small molecule activator, from a common pathogenic mutation of g6pdx. In order to develop a suitable therapeutic agent, the mechanism of the molecule, AG1, was studied by the group. They postulated that AG1 promoted G6PD dimerization. This was achieved by the bridging of two NADP+ binding sites. The C2-symmetric region of the G6PD homodimer is important for activation which is targeted by AG1.44 Due to the observed increase in activity of enzyme by AG1, it can be considered in the treatment of G6PD deficiency. This drug can also be used in the prevention of complications that arise as a consequence of the deficiency.45 However, it was observed that AG1 was not able to activate the enzyme in certain kinds of mutations. AG1 still is a novel class molecule that promotes activation in multiple pathogenic mutants.44
G6PD deficiency is a common enzymatic disorder that is inherited in an X-linked manner. Unlike other enzymatic disorders, G6PD is not rare; however, the majority of the population affected by the disease are asymptomatic, unless they inherit a severe mutant copy of the enzyme. Clinical features appear only after exposure to certain oxidative stressors which primarily include antimalarial drugs and fava beans. Diagnostic measures to detect the presence of G6PD deficiency include enzymatic activity detection and molecular analysis. Newer techniques have been formulated in the past few years that have demonstrated better efficacy in diagnosing the condition compared to existing techniques. Currently, there is no effective treatment for the deficiency, removal of the trigger is frequently all that is required. In certain cases, blood transfusion and haemodialysis may be necessary.
Neonatal jaundice is a common early clinical manifestation of the disease. In the recent past, phototherapy has been used as a prophylactic measure for the management of the condition, along with avoidance of offending oxidative stressors. Various studies have been conducted to prove the effectiveness of the therapy and, although there are serious adverse reactions that have been reported due to phototherapy, its therapeutic effect outweighs the risk. Pharmacological modulation of the interactome is the future in drug discovery in this area. It is known that PPIs are important in enzyme stability and activation along with cell signalling. Hence, interventions acting on this target will likely prove immensely valuable in drug discovery, particularly for G6PD deficiency. Treatment strategies for the deficiency have seen significant advances in recent years, with small molecule activators demonstrating effective increases in enzymatic activity, albeit in in vitro studies. Further research needs to be conducted in this area to develop these small molecule activators for the treatment of G6PD deficiency.
None.
The Authors have no funding, financial relationships or conflicts of interest to disclose.
Writing – Original Draft: NR, Writing – Review and Editing: NR, GG, Visualization: NR, Supervision: GG
1. National Organization for Rare Disorders. Glucose-6-Phosphate Dehydrogenase Deficiency. Available from: https://rarediseases.org/rare-diseases/glucose-6-phosphate-dehydrogenase-deficiency. Cited: June 1, 2020.
2. Lo E, Zhong D, Raya B, Pestana K, Koepfli C, & Lee MC. Prevalence and distribution of G6PD deficiency: implication for the use of primaquine in malaria treatment in Ethiopia. Malar J. 2019 Oct 7;18:340.
3. Belfield KD, Tichy EM. Review and drug therapy implications of glucose-6-phosphate dehydrogenase deficiency. Am J Health Syst Pharm. 2018 Feb 1;75(3):97-104.
4. Lee MH, Malloy CR, Corbin IR, Li J, Jin ES. Assessing the pentose phosphate pathway using [2, 3-13 C2] glucose. NMR Biomed. 2019 Mar 29;32(6):e4096.
5. Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev. 2007 Sep;21(5):267-83.
6. Drousiotou A, Touma EH, Andreou N, Loiselet J, Angastiniotis M, Verrelli BC, Tishkoff SA. Molecular characterization of G6PD deficiency in Cyprus. Blood Cells Mol Dis. 2004 Jul-Aug;33(1):25-30.
7. Glucose-6-phosphate dehydrogenase deficiency. WHO Working Group. Bull World Health Organ. 1989;67(6):601-611.
8. Nkhoma E, Poole C, Vannappagari V, Hall S, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: A systematic review and meta-analysis. Blood Cells Mol Dis. May-Jun 2009;42(3):267-78.
9. Harcke SJ, Rizzolo D, Harcke HT. G6PD deficiency: An update. JAAPA. 2019 Nov;32(11):21-26.
10. Brewer GJ, Tarlov AR, Alving AS. The methemoglobin reduction test for primaquine-type sensitivity of erythrocytes. A simplified procedure for detecting a specific hypersusceptibility to drug hemolysis. JAMA. 1962 May 5;180:386-8.
11. Luzzatto L, Seneca E. G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J Haematol. 2014 Feb;164(4):469-80.
12. Hecker PA, Leopold JA, Gupte SA, Recchia FA, Stanley WC. Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013 Feb 15;304(4):H491-500.
13. Finley, D. S. Glucose-6-phosphate Dehydrogenase Deficiency Associated Stuttering Priapism: Report of a Case. J Sex Med. 2008 Dec 3;5(12):2963-2966
14. Naylor CE, Rowland P, Basak AK, Gover S, Mason PJ, Bautista JM, Vulliamy TJ, Luzzatto L, Adams MJ. Glucose 6-phosphate dehydrogenase mutations causing enzyme deficiency in a model of the tertiary structure of the human enzyme. Blood. 1996 Apr 1;87(7):2974-82.
15. Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008 Jan 5;371(9606):64-74.
16. Roper D, Layton M, Rees D, Lambert C, Vulliamy T, De la Salle B, et al. British Society for Haematology. Laboratory diagnosis of G6PD deficiency. A British Society for Haematology Guideline. Br J Haematol. 2020 Apr;189(1):24-38.
17. Gómez-Manzo S, Marcial-Quino J, Ortega-Cuellar D, Serrano-Posada H, González-Valdez A, Vanoye-Carlo A, et al. Functional and Biochemical Analysis of Glucose-6-Phosphate Dehydrogenase (G6PD) Variants: Elucidating the Molecular Basis of G6PD Deficiency. Catalysts. 2017 May 2;7(135).
18. Persico MG, Viglietto G, Martini G, Toniolo D, Paonessa G, Moscatelli C, et al. Isolation of human glucose-6-phosphate dehydrogenase (G6PD) cDNA clones: primary structure of the protein and unusual 5' non-coding region. Nucleic Acids Res. 1986 Mar 25;14(6):2511-22.
19. Doss CG, Alasmar DR, Bux DI, Sneha P, Bakhsh FD, Al-Azwani I, et al. Genetic epidemiology of glucose-6-phosphate dehydrogenase deficiency in the Arab World. Sci Rep. 2016;6:37284.
20. Sarker SK, Islam MT, Eckhoff G, Hossain MA, Qadri SK, Muraduzzaman AK, et al. Molecular Analysis of Glucose-6-Phosphate Dehydrogenase Gene Mutations in Bangladeshi Individuals. PLoS One. 2016 Nov 23;11(11):e0166977.
21. Beutler E. G6PD deficiency. Blood. 1994 Dec 1;84(11):3613-36.
22. Beutler E, Blume KG, Kaplan JC, Löhr GW, Ramot B, Valentine WN. International Committee for Standardization in Haematology: recommended screening test for glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Br J Haematol. 1979 Nov;43(3):465-7.
23. Tantular IS, Kawamoto F. An improved, simple screening method for detection of glucose-6-phosphate dehydrogenase deficiency. Trop Med Int Health. 2003 Jun;8(6):569-74.
24. Bancone G, Kalnoky M, Chu CS, Chowwiwat N, Kahn M, Malleret B, et al. The G6PD flow-cytometric assay is a reliable tool for diagnosis of G6PD deficiency in women and anaemic subjects. Sci Rep. 2017 Aug 29;7(1):9822.
25. Adan A, Alizada G, Kiraz Y, Baran Y, Nalbant A. Flow cytometry: basic principles and applications. Crit Rev Biotechnol. 2017 Mar;37(2):163-176.
26. Bancone G, Gornsawun G, Chu CS, Porn P, Pal S, Bansil P, et al. Validation of the quantitative point-of-care CareStart biosensor for assessment of G6PD activity in venous blood. PLoS One. 2018 May 8;13(5):e0196716.
27. Ley B, Alam MS, O'Donnell JJ, Hossain MS, Kibria MG, Jahan N, et al. A Comparison of Three Quantitative Methods to Estimate G6PD Activity in the Chittagong Hill Tracts, Bangladesh. PLoS One. 2017 Jan 25;12(1):e0169930.
28. Minucci A, Gentile L, Zuppi C, Giardina B, Capoluongo E. Rapid and simple identification of the commonest glucose-6-phosphate dehydrogenase (G6PD) Italian mutations: from DNA extraction to genotyping. Clin Chim Acta. 2012 Jun 14;413(11-12):1018-9.
29. Nemours KidsHealth. G6PD Deficiency, Available from: https://kidshealth.org/en/parents/g6pd.html. Last updated July 2018; cited: February 20, 2020.
30. Tan KL. Phototherapy for neonatal jaundice in erythrocyte glucose-6-phosphate dehydrogenase-deficient infants. Pediatrics. 1977 Jun;59 Suppl(6 Pt 2):1023-6.
31. Faulhaber FRS, Procianoy RS, Silveira RC. Side Effects of Phototherapy on Neonates. Am J Perinatol. 2019 Feb;36(3):252-257
32. Stokowski LA. Fundamentals of phototherapy for neonatal jaundice. Adv Neonatal Care. 2011 Oct;11(5 Suppl):S10-21.
33. Itoh S, Okada H, Kuboi T, Kusaka T. Phototherapy for neonatal hyperbilirubinemia. Pediatr Int. 2017 Sep;59(9):959-966.
34. Xiong T, Qu Y, Cambier S, Mu D. The side effects of phototherapy for neonatal jaundice: what do we know? What should we do? Eur J Pediatr. 2011 Oct;170(10):1247-55.
35. Csoma Z, Hencz P, Orvos H, Kemeny L, Dobozy A, Dosa-Racz E, et al. Neonatal blue-light phototherapy could increase the risk of dysplastic nevus development. Pediatrics 2007;119(05):1036-1037
36. Speck WT, Chen CC, Rosenkranz HS. In vitro studies of effects of light and riboflavin on DNA and HeLa cells. Pediatr Res. 1975 Mar;9(3):150-3.
37. Ballowitz L, Bunjamin A, Hanefeld F, Lietz L, Stüttgen G, Wirjadi D. Effects of riboflavin on Gunn rats under phototherapy. Pediatr Res. 1979 Dec;13(12):1307-15.
38. Ennever, J., Speck, W. Photodynamic Reaction of Riboflavin and Deoxyguanosine. Pediatr Res. 1981 Jun 1;15(1):956-958.
39. Ennever JF, Speck WT. Photochemical reactions of riboflavin: covalent binding to DNA and to poly (dA). poly (dT). Pediatr Res. 1983 Mar;17(3):234-6.
40. Wickremasinghe AC, Kuzniewicz MW, Grimes BA, McCulloch CE, Newman TB. Neonatal Phototherapy and Infantile Cancer. Pediatrics. 2016 Jun;137(6):e20151353.
41. Newman TB, Wickremasinghe AC, Walsh EM, Grimes BA, McCulloch CE, Kuzniewicz MW. Retrospective Cohort Study of Phototherapy and Childhood Cancer in Northern California. Pediatrics. 2016 Jun;137(6):e20151354.
42. Le TN, Reese J. Bronze Baby Syndrome. J Pediatr. 2017 Sep;188:301-301.e1.
43. Kopelman AE, Brown RS, Odell GB. The “bronze” baby syndrome: a complication of phototherapy. J Pediatr. 1972 Sep;81(3):466-72.
44. Raub AG, Hwang S, Horikoshi N, Cunningham AD, Rahighi S, Wakatsuki S, Mochly-Rosen D. Small-Molecule Activators of Glucose-6-phosphate Dehydrogenase (G6PD) Bridging the Dimer Interface. ChemMedChem. 2019 Jul 17;14(14):1321-1324.
45. Hwang S, Mruk K, Rahighi S, Raub AG, Chen CH, Dorn LE, et al. Correcting glucose-6-phosphate dehydrogenase deficiency with a small-molecule activator. Nat Commun. 2018 Oct 2;9(1):4045.
Nidhruv Ravikumar, 1 Medical student, Queen's University Belfast, Northern Ireland, UK
Graeme Greenfield, 2 BA, BM, BCh, MRCP. Wellcome Trust, Queen's University Belfast, Belfast City Hospital, Northern Ireland, UK
About the Author: Nidhruv Ravikumar is currently a 3rd year medical student at Queen's University Belfast of a 5-year program in MBBCh BAO. He is enthusiastically involved in research in various fields of medicine, and keeps himself well-informed with latest advancements in the field. He is also an active member of various medical societies and is the Vice-President of the Asian Medical Students Association Northern Ireland.
Correspondence: Nidhruv Ravikumar. Address: University Rd, Belfast BT7 1NN, United Kingdom. Email: nravikumar01@qub.ac.uk
Editor: Paul MacDaragh Ryan Student Editors: Yusuff Adebisi & Brandon Belbeck Copyeditor: Leah Komer Proofreader: Leah Komer Layout Editor: Annora A. Kumar
Cite as: Ravikumar N, Greenfield G. Glucose-6-phosphate Dehydrogenase Deficiency: A Review. Int J Med Students. 2020 Sep-Dec;8(3):281-7.
Copyright © 2020 Nidhruv Ravikumar, Graeme Greenfield
This work is licensed under a Creative Commons Attribution 4.0 International License.
International Journal of Medical Students, VOLUME 8, NUMBER 3, December 2020