CT Angiography of Abdomen
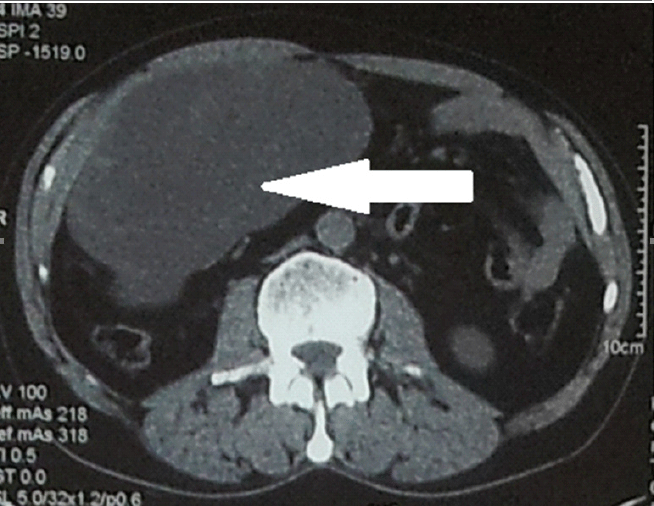
Axial or transverse section of abdomen with a large sarcomatoid lesion on the right side (white arrow)
Stefan Krstevski1
doi: http://dx.doi.org/10.5195/ijms.2016.164
Volume 4, Number 3: 120-122
Received 30 12 2015: Accepted 07 12 2016
ABSTRACT
Background:Gastrointestinal stromal tumor (GIST) is a rare gastrointestinal tumor but the most common mesenchymal tumor. It originates in the interstitial cells of Cajal, which on their own membranes have a specific antigen designated as CD117 or KIT receptor, a reliable indicator of tumor detection and therapy.
Case:We reported a case of a 63-year-old male patient, who presented with symptoms of severe abdominal pain, discomfort, and secondary anemia. After a physical examination and a CT scan of the abdomen with angiography, the presence of a large tumor in the right hypochondrium was detected. Blood tests showed a high C-reactive protein and low hemoglobin. The patient had an exploratory laparotomy on his abdomen with complete removal of the tumor growth, which was then histologically analyzed. The obtained result from the histological analysis showed a high-degree of GIST.
Conclusion:Early diagnosis of GISTs is of paramount importance to reduce mortality rate. This requires good background in pathobiology and knowledge of the physical manifestations of this malignant tumor. When this is combined with various imaging techniques, it becomes the gold standard method for GIST diagnosis.
Keywords: Gastrointestinal Stromal Tumors; Abdominal Pain; Laparotomy; Early Diagnosis.
Gastrointestinal stromal tumor (GIST) represents only 1-3% of all gastrointestinal tumors but is the most common mesenchymal tumor, with a prevalence of 80%.1 This tumor originates in the interstitial cells of Cajal (ICC). These interstitial cells are innervated by the peripheral nerves and placed in the plexus mesentericus, thus functioning as pacemakers to determine the motility rhythm of the digestive tract.2-4 GIST is asymptomatic in 35% of clinical cases, while in other cases it manifests as abdominal pain, discomfort (heavy sensation as if having a foreign body in the abdomen), bleeding, and secondary anemia.5 As for its localization, it may be located in the digestive tract or outside it, as an extra GIST in the abdominal pelvic cavity, omentum, mesenterium, uterus, or retroperitoneally.2,6 It is localized in the stomach in 60–70% of the cases, followed by small intestine in 20–25% of cases, colorectal in 5% of cases, and esophageal in 5% of cases.7
The incidence of GIST has increased over time from 0.028 per 100,000 in 1992 to 0.688 per 100,000 in 2002, and affects 7–14 people in a million at present.7 Statistics show that there is no significant difference between sexes, and the male-to-female ratio is 1:1.7 The usual age when GIST is diagnosed is between 55 and 60 years.
GIST is a challenge for modern medicine and research since it has a late manifestation due to the long latency period. It is also of great concern for medical fellows since it is increasingly present in gastrointestinal pathology with rather high rate of mortality. It is therefore important to ensure a timely diagnosis, followed by chemotherapy and/or minimally-invasive surgical treatment.
For a long period of time, GIST was considered a type of leiomyoma or neuroma, until immuno-histochemical staining techniques revealed that the tumor is a specific type of mesenchymal tumor. The ICC cells from which it derives are cells that have a specific antigen on their own membrane, designated as CD117 or KIT tyrosine kinase receptor. This is present in over 85% of these tumors, which calls for the recommended monoclonal anti-KIT therapy (imatinib and sunitinib).2,8,9 Furthermore, in 3–5% of GIST, there is a positive platelet derived growth factor subunit A (PDGFA).6 This receptor shows exact same characteristics as KIT. There is also a third form of GIST expression called the wild type, which is immuno-histochemically positive on insulin-like growth factor 1 receptor (IGF1R). Microscopically, the GIST can occur in three forms: spindle cells (the most common type: 70%), epitheloid cells (20%) and mixed (10%).10 Macroscopically, GIST is characterized by multi-nodularity, where the surface may be either smooth or granular, with zones of hemorrhage, necrosis, calcification and cystic changes.10
A 63-year-old male presented with abdominal pain and discomfort in the lower right abdominal quadrant with propagation towards the right groin. The symptoms, though less severely, appeared eight months before his hospitalization. The pain was treated with analgesics, and echo-tomography of the abdomen was made, but no visible pathological changes were found. Month by month, his pain and discomfort grew in intensity, yet the patient did not seek any medical attention. Right before his hospitalization, the patient suffered another pain attack so high in intensity that knocked him unconscious. He was immediately hospitalized in the clinic, where a physical examination of the abdomen was performed. With a deep palpation in the right hypochondrium, a large-size tumor which was insensitive to pain was palpated, whereas the liver and spleen were impalpable. Laboratory blood tests showed low level of hemoglobin (99 g/L) indicating secondary anemia, and the urine analysis showed decreased lactate dehydrogenase (LDH) (179 U/L; normal range = 213–423 U/L) and increased C-reactive protein (CRP) (23.6 mg/L; normal range = 0.0–5.0 mg/L), indicating an active inflammation.
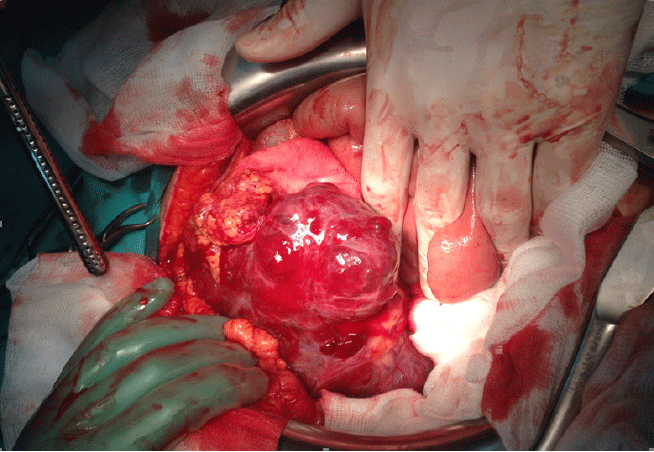
CT angiography of abdomen showed the following results: tumor mass in right hemiabdomen, sub-hepatic, next to the hepatic flexure medial of ceco-ascendens and extends distally almost to the right iliac fossa. The tumor mass was visibly re-stained with a stronger contrast. It was attached to a large sarcomatoid mass with mixed interiority, without significantly increased LGL (Figure 1). Additional medical check-up was conducted, but no other pathological changes were found. The patient weighed 161 pounds (73 kg), and his blood pressure was 135/90 mmHg. Based on the results, the diagnosis was Tu. abdominis sang. Haemascos, Anemia sec. A laparotomy exploration was then performed to remove the neoplastic tissue in its entirety and to further subject it to histological analysis. Medial laparotomy was performed operationally with hemoperitoneum findings revealing a bleeding cystic type of neoplasm in the right hypochondrium (Figure 2). Extirpation of the neoplasm was done in full. The patient’s stomach was deserosated, after which it was serosated with more individual stitches. Profuse lavage followed, and upon determining exact hemostasis, two drains were placed—one subhepatally and one in rectovesical excavation. The surgical wound was closed in layers. The entire surgical treatment was done with general endotracheal anesthesia. The tissue material was sent to the pathology laboratory for ultimate histological analysis. Fourteen pieces of paraffin were taken for analysis.
Figure 1.CT Angiography of Abdomen
Hemorrhagic Cystic Type of Neoplasm Was Found in the Right Hypochondrium
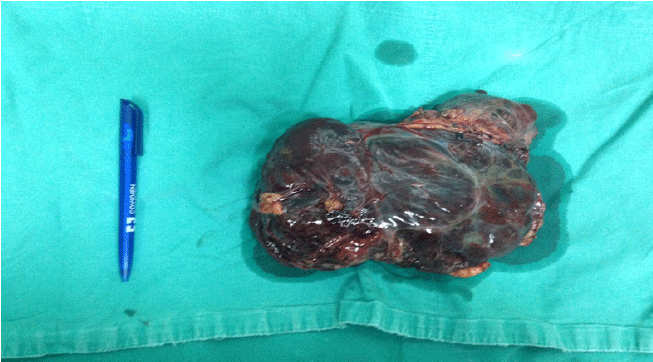
The tumor mass was 18 cm × 11 cm × 5 cm in size and weighed 1.5 pounds (675 g) (Figure 3). A thin connective capsule was visible on the surface. At the intersection, the tumor tissue had a sponge-like appearance with multi-cystic degeneration and bleeding areas.
Figure 3.Macroscopic Finding of Tumor Mass
Microscopic analysis showed neoplastic proliferation embedded with epithelial cells in solid arrangement. Magnification of the cells shows eosinophilic and bright cytoplasm with oval or elongated nuclei. In some of these nuclei, nucleoli were also clearly visible. The cells were pleomorphic with manifested nuclei atypia. The tumor also exhibited blood vessels, edema, and chronic inflammation. An immuno-histochemical method was employed to prove the origin of the tumor by using CD117, CD34, actin, Vimetin, C-100, CKWS, Melan A, and HMB 45. Strong positivity of CD117 and CD34 was evident, whereas actin was positive in only 10%. Proliferative findings of Ki-67 were <10%. Mitotic figures were >5/50 HPF, as per usual. This finding suggests gastrointestinal stromal tumor, with a high mitotic index, pT4. Since a high-risk GIST was established, the patient was sent to the Oncology Clinic for further treatment. The oncological treatment included chemotherapy with imatinib, a specific monoclonal antibody for tyrosine kinase receptor, in a dosage of 400 mg twice a day. The follow-up imaging was to be done every six months during the adjuvant therapy, every 3–4 months in the first two years after the received chemotherapy, and every 6–12 months for up to 10 years after the surgery.
Reaching the diagnosis of GIST is mainly based on imaging procedures and histological analysis with immuno-histochemical staining. Imaging procedures such as endoscopy, CT, MRI and PET-scan are needed to determine the localization and size of tumor size as well as monitor the effectiveness of therapy and relapse of illness. Histological analysis is used to determine the distinct characteristics and nature of the tumor tissue through immuno-histochemical staining and the use of anti-CD117 antibodies to prove the presence of KIT receptor.5,7
The classification of GIST is based on the Risk Stratification System of primary GIST, which determines the risk of recurrence and metastasis and indirectly informs the planning of therapy and patient survival.6 The prognostic models used in this system include variables such as size, localization, and mitotic index cutoff of 5/50 HPF to stratify patients into very low, low, medium and high risk groups (very low risk: 0%, low: 3–9%, medium: 12–24%, high: 55–90% with a possibility of recurrence and metastasis and inversely proportional to that, of survival).7
The GISTs are also characterized by metastasis that commonly occurs in the abdominal cavity and liver; they can be found infrequently in the soft tissues and bones as well.6,10 But typical of GIST is that it does not metastasize to the lymph nodes, i.e. its pattern of metastasis is exclusively hematogenous. A series of studies show that the median survival in patients with GIST is 60 months when no metastases are found. In cases when the tumor metastasizes, the chance of survival is 12–19 months.1
Early diagnosis of GIST is of crucial importance to reduce mortality rate among affected patients. This requires good knowledge of pathobiology and the characteristics of this tumor, especially of the KIT receptor that is GIST-specific. Immuno-histochemical staining has high specificity and sensitivity and contributes to a reliable diagnosis of the GIST. This technique combined with different imaging methods such as CT, MRI, ECHO, and PET SCAN is the gold standard for tumor diagnosis.
Key Points:Herein, I would like to express my deep sense of gratitude to my professor and mentor Zoran Karadzov, MDCM, as well as to Darko Dzambaz, MD, both associates at the University Clinic of Digestive Surgery – Skopje, for their enthusiastic support, guidance and counseling. In addition, I express my gratitude to the male patient for his trust and support over the critical period of his tumor surgical treatment. Finally, I also acknowledge the kind support and advice of my fellow medical student, Elena Peneva.
The author has no funding, financial relationships, or conflicts of interest to disclose.
Conceptualization, Writing: SK.
1. Nowain A, Bhakta H, Pais S, Kanel G, Verma S. Gastrointestinal stromal tumors: clinical profile, pathogenesis, treatment strategies and prognosis. J Gastroenterol Hepatol. 2005 Jun;20(6):818–24.
2. Min KW, Leabu M. Interstitial cells of Cajal (ICC) and gastrointestinal stromal tumor (GIST): facts, speculations, and myths. J Cell Mol Med. 2006 Oct-Dec;10(4):995–1013.
3. Ward SM, Ordog T, Bayguinov JR, Horowitz B, Epperson A, Shen L, et al. Development of interstitial cells of Cajal and pacemaking in mice lacking enteric nerves. Gastroenterology. 1999 Sep;117(3):584–94.
4. Sanders KM, Ward SM. Interstitial cells of Cajal: a new perspective on smooth muscle function. J Physiol. 2006 Nov 1;576(Pt 3):721–6.
5. Nishida T, Blay JY, Hirota S, Kitagawa Y, Kang YK. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer. 2016 Jan;19(1):3–14.
6. Tan CB, Zhi W, Shahzad G, Mustacchia P. Gastrointestinal stromal tumors: a review of case reports, diagnosis, treatment, and future directions. ISRN Gastroenterol. 2012;2012:595968.
7. Eisenberg BL, Pipas JM. Gastrointestinal stromal tumor–background, pathology, treatment. Hematol Oncol Clin North Am. 2012 Dec;26(6):1239–59.
8. Isozaki K, Hirota S. Gain-of-function mutations of receptor tyrosine kinases in gastrointestinal stromal tumors. Curr Genomics. 2006;7(8):469–75.
9. Yan L, Zou L, Zhao W, Wang Y, Liu B, Yao H, et al. Clinicopathological significance of c-KIT mutation in gastrointestinal stromal tumors: a systematic review and meta-analysis. Sci Rep. 2015 Sep 9;5:13718.
10. Raghavan D, Blanke CD, Johnson DH, Moots PL, Reaman GH, Rose PG, et al. Textbook of uncommon cancer. 4th ed. New Jersey: Wiley; 2012.
Stefan Krstevski, 1 Faculty of Medicine, Saints Cyril and Methodius University of Skopje, Skopje, Republic of Macedonia.
About the Author: Stefan Krstevski is a sixth-year medical student at Medical Faculty-Skopje R.Macedonia who is interested in the field of Oncology Surgery.
Correspondence Stefan Krstevski. Address: Faculty of Medicine, Saints Cyril and Methodius University of Skopje, Skopje 1000, Republic of Macedonia. Email: stefan.krstevski@ymail.com
Cite as: Krstevski S. Gastrointestinal stromal tumor: a case report. Int J Med Students. 2016 Sep-Dec;4(3):120-2.
Copyright © 2016 Stefan Krstevski
International Journal of Medical Students, VOLUME 4, NUMBER 3, December 2016