Case Report
The Importance of Rapid Consideration of Creutzfeldt-Jakob Disease in the Differential
Diagnosis of Progressive Neurodegenerative Disease: A Case Report
Arthur Joseph1, Jacob Core1, Daniel Solano1, Marquand Patton2, Shaun Smart2
doi: http://dx.doi.org/10.5195/ijms.2016.154
Volume 4, Number 2: 72-75
Received 13 10 2015:
Accepted 01 05 2016
ABSTRACT
Background:
Creutzfeldt-Jakob disease (CJD) is a prion disease characterized by misfolded proteins
that lead to neurodegeneration and inevitable death. Classic sporadic CJD presents
primarily with cognitive symptoms and ataxia without visual impairment at the onset
of the illness. Seizure activity is a rare presentation of patients with sporadic
CJD.
Case:
We present a rare case of rapidly progressive encephalopathy in a 57-year-old female
who presented to the emergency department with bizarre behavior and vision deterioration.
Imaging was unrevealing, and infectious and organic causes were ruled out. Electroencephalogram
showed evidence of encephalopathy and non-convulsive status epilepticus. Magnetic
resonance imaging conducted later displayed high signal intensity in centrum ovale.
The patient’s history, results from diagnostic analyses, and clinical presentation
suggested the diagnosis of CJD (sporadic type).
Conclusion:
Due to the low incidence and varying clinical presentations, it is difficult to include
CJD in a differential diagnosis without specific analytic measures. However, for the
benefit of the patient and healthcare resources, CJD needs to be quickly considered
when rapid neurological decline or non-convulsive status epilepticus is not suggestive
of another entity.
Keywords:
Creutzfeldt-Jakob Syndrome;
Status Epilepticus;
Diagnosis.
Introduction
Creutzfeldt-Jakob disease (CJD), the most iconic of the human prion pathologies, is
characterized by misfolded proteins that lead to neurodegeneration and inevitable
death. CJD displays an exceedingly fast rate of deterioration, with a median time
from onset of symptoms to death of 12 months. The time from onset of symptoms to diagnosis
varies, with a median of approximately 6 months.1 With an estimated incidence of one to two per million people annually, CJD is a rare
but important consideration in the differential diagnosis of rapid neurodegeneration.2,3
Four major types of CJD exist: iatrogenic, variant, familial, and sporadic CJD.2 Sporadic CJD (sCJD) is responsible for 85% of CJD.3,4 Compared to other CJDs, sCJD has been shown to occur at a later age and affect women
more frequently than men.5 Symptoms of sCJD include personality changes, sleep disturbances, cognitive function
decline, behavioral abnormalities, visual abnormalities, hallucinations, cerebellar
dysfunction, memory decline, myoclonus, progressive dementia, a positive startle reflex,
and pyramidal dysfunction.5
Several subtypes of sCJD have been studied. Classic sCJD has been described primarily
with cognitive symptoms and ataxia without visual impairment at the onset of the illness.
Classic sCJD notoriously has a short interval between symptom onset and diagnostic
evaluation.3 Seizure activity is a rare presentation in sCJD, and a review of the literature up
to 2010 found that 12 patients have been reported to present with CJD and nonconvulsive
status epilepticus (NCSE).6 In this report, we present a rare case of rapidly progressive encephalopathy in a
57-year-old female, who presented with cognitive decline and NCSE, which lead to the
diagnosis of sCJD. All appropriate consents, including the use of this case for educational
or research purposes, were obtained from the patient prior to admission.
The Case
A 57-year-old Hispanic female presented to our facility for an involuntary psychiatric
evaluation for depression. The patient appeared disoriented and confused, with limited
response to verbal/non-verbal cues and a flattened affect. According to the patient’s
spouse, the symptoms began three months prior to presentation with changes in vision
and a decrease in responsiveness. Symptoms progressed, and the patient eventually
had difficulty maintaining her employment. The spouse described her as having an unfocused
gaze that fluctuated, difficulty with coordination, personality changes, and a decline
in activities of daily living. She spent most of her day in silence. The day after
her admission, neurology specialist was consulted for the patient’s altered mental
status, and a full physical exam was limited by the patient’s declining health. The
initial computed tomography (CT) scan and magnetic resonance imaging (MRI) scan of
her brain were unremarkable. An electroencephalogram (EEG) was also ordered to rule
out an epileptic cause for the patient’s episodes of unresponsiveness. Before the
tests were administered, the patient was reported to have uncontrolled arm movements
that prompted a transfer to the Emergency Room at Palmetto General Hospital. In the
emergency room, tests for rapid plasma reagin (RPR), B12, folate, thyroid stimulating
hormone (TSH), and ammonia levels were ordered. At this point, the patient was still
mildly alert and able to follow simple commands. She did, however, exhibit bizarre
behavior, including anger, anxiety and irritability.
The video EEG was very concerning, as it showed symmetric/reactive background slowing
activity at 5 Hz with intermittent triphasic morphology consistent with moderate-to-severe
encephalopathy, as well as NCSE. Subsequent EEGs were consistent with these findings,
and treatment was attempted with phenytoin (Dilantin). After anticonvulsant therapy
failed to reverse or slow the rate of her neurodegeneration, a lumbar puncture was
ordered for the analysis of 14-3-3 and tau protein for a suspected rapidly progressing
encephalopathy. After a week of deteriorating and inconclusive laboratory analysis,
the patient’s history of gastric band surgery prompted further laboratory tests. Her
thiamine level was below normal limits, which prompted concern for the possibility
of Wernicke or Korsakoff encephalopathy. Thiamine levels were replaced without an
improvement in neurological function. Repeat EEGs continued to show signs of encephalopathy
with generalized sharp wave discharges that appeared to be epileptiform in nature
(Figure 1). While awaiting the results of 14-3-3 and tau protein immunoassay, other laboratory
tests returned negative results, including the tests for human immunodeficiency virus
(HIV) and herpes simplex virus (HSV). At this point, the differential diagnosis was
almost completely narrowed down to CJD. Autoimmune encephalitis, mitochondrial disease,
heavy metal toxicity, paraneoplastic panel, derangements in amino acids, and organic
acids and carnitine abnormalities were all ruled out.
Figure 1.
Repeat Electroencephalogram of the Patient
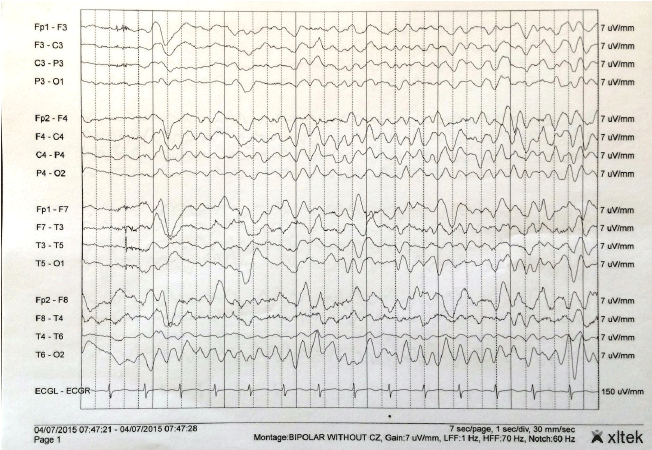
Electroencephalogram (EEG) of the patient displaying frequent triphasic spike and
wave complexes. Discharges were frequent and at times continuous, lasting for hours.
EEG was reviewed in a double banana montage with sensitivity at 7 microvolts and a
time base of 30 mm/s. Background activity was seen primarily at 4–5 Hz.
Several days later, a repeat MRI revealed a single area of hyper-intensity involving
the right centrum semiovale consistent with a non-specific white matter lesion (Figure 2). The cerebrospinal fluid (CSF) was positive for 14-3-3, and tau protein immunoassay
revealed a tau protein level of 6,350 mg/dL. The patient’s clinical presentation,
along with supporting results from MRI, EEG, 14-3-3 and tau protein immunoassay, all
supported the diagnosis of CJD. While the definitive diagnosis was only achieved post-mortem,
the findings were discussed with an expert in prion disease at the University of Chicago,
who agreed with the likely diagnosis of CJD. The family, once informed of the findings,
gave consent to genetic and post-mortem analysis. The patient later received hospice
care, and the National Prion Surveillance Center had been informed of impending autopsy
for genetic analysis.
Figure 2.
Brain Magnetic Resonance Imaging (MRI) of the Patient
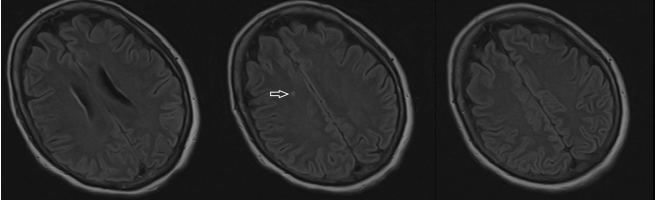
Three fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI)
axial slices of the brain without gadolinium. Arrow: Single area of localized hyperintensity
involving the right centrum semiovale above the lateral ventricles.
Discussion
The patient presented with severe neurological dysfunctions as demonstrated by her
disorientation, confusion, flat affect, and limited responsiveness. However, retrospective
questioning revealed that while these symptoms brought her to the hospital, other
symptoms had been present for months. This considerable length of time is significant
in that it represents a large portion of the total duration from disease onset to
eventual death. Furthermore, CJD was only considered after the EEG demonstrated refractory
NCSE and reversible causes of CJD had been ruled out, adding to the delay in probable
diagnosis.
NCSE is an atypical finding in cases of sCJD, with only 12 cases being reported prior
to this.6 Patients displaying this symptomatology were treated as having refractory NCSE and
were non-responsive to multiple doses of antiepileptic drugs. Our patient was also
unresponsive to several doses of phenytoin (Dilantin), which prompted the workup for
sCJD.
The early stages of sCJD are largely nonspecific, making it difficult to suspect early
on. As described earlier, subtle early symptoms include personality changes, sleep
disturbances, and visual impairment. The classic finding of myoclonic jerks is typically
seen later in the disease course and may be the eventual trigger for the sCJD workup.5
The diagnostic criteria for CJD outlined by the World Health Organization and Centers
for Disease Control and Prevention (CDC) classify CJD into either definite, probable,
or possible based on symptomatology, EEG findings, CSF analysis, and MRI (Table 1). Definitive diagnosis of CJD requires a tissue biopsy and is confirmed post mortem.
However, updated criteria which may be superior to the current standard have been
proposed.7
Table 1.
Diagnostic Criteria for Sporadic Creutzfeldt-Jakob Disease (sCJD).
| Possible Case |
- Progressive dementia; and
- Atypical EEG; and
- <2 year duration; and
- At least two of the following:
- Myoclonus
- Visual or cerebellar disturbance
- Pyramidal or extra pyramidal dysfunction
- Akinetic mutism
|
| Probable Case (in the absence of alternative diagnosis) |
- Progressive dementia;
- Two of the four clinical features mentioned above for possible case, with
- A typical EEG of generalized triphasic periodic complexes at the rate of one per second;
and/or
- Positive 14-3-3 assay in CSF; and
- MRI showing high signal abnormalities in the caudate nucleus and/or putamen on diffusion-weighted
imaging (DWI) or fluid-attenuated inversion recovery (FLAIR).
- Akinetic mutism
|
| Definite Case |
- Neuropathological confirmation post-mortem; and/or
- Presence of protease-resistant prion protein by either Western Blot or immunochemistry;
and/or
- Presence of scrapie associated fibrils.
|
Diagnostic evaluation of patients with suspected sCJD involves assessment of the 14-3-3
protein assay, MRI, and EEG. Studies have demonstrated that CSF 14-3-3 may be the
most accurate test, with a sensitivity of 92% and a specificity of 80%.8 Typical EEG findings of sCJD show periodic sharp and slow wave complexes (PSWC) with
a reported sensitivity and specificity of 66% and 74%, respectively.8 Our patient demonstrated these changes; however, wave complexes consistent with NCSE
were also present. sCJD mimicking NCSE on EEG is extremely rare, as only 12 prior
cases of CJD masquerading as NCSE have been documented.6 The importance of such finding has yet to be determined. MRI has shown to be the
superior test by demonstrating areas of hyperintensity in the cortical and/or subcortical
areas with sensitivity and specificity of 92.3% and 95%, respectively.8,9 Multiple studies found MRI to be superior in the evaluation of patients with suspected
sCJD compared to EEG and 14-3-3 protein.9 With the demonstrated sensitivity and specificity of diffusion-weighted imaging (DWI)
and fluid-attenuated inversion recovery (FLAIR) for white matter disease, MRI has
become essential in the evaluation of rapidly progressive dementia.9
The National Prion Disease Pathology Surveillance Center (NPDPSC) provides CSF analysis
for tau protein levels and the existence of 14-3-3 protein through real-time quaking-induced
conversion (RT-QuiC) for the detection of abnormal prion protein. The center has reported
a 98.5% specificity and up to 92% sensitivity in CSF analysis of 193 cases that have
been autopsy-verified by the NPDPSC (Available from: http://case.edu/med/pathology/centers/npdpsc/index.html; cited 2015 May 31).
The RT-QuiC is reported as positive or negative. As reported by the NPDPSC, tau protein
levels of 0–899 pg/mL correspond with a 25% probability of prion disease, 900–1,149
pg/mL with a 36% probability, and greater than 1,150 pg/mL with a 76% probability
(Available from: http://case.edu/med/pathology/centers/npdpsc/index.html; cited 2015 May 31). Our patient was positive for RT-QuiC and 14-3-3 protein and
had a t-tau protein level of 6,350 pg/mL. After careful review of the literature,
the consensus appears to be in agreement with the data reported by the NPDPSC. In
fact, one study of 100 patients with varying neurodegenerative diseases found that
significantly elevated levels of t-tau protein was found in CJD alone (CJD and variant
CJD) (Available from: http://www.who.int/zoonoses/diseases/Creutzfeldt.pdf; cited 2015 Jun 25).
Conclusion
Ruling out reversible encephalopathic disease is important and potentially life-saving,
but can be challenging and time consuming. CJD, particularly the sporadic subtype,
is a devastating disease with a rapid neurological degeneration pattern. Due to the
low incidence and varying clinical presentations, it is difficult to include CJD as
a differential diagnosis without specific and supportive analytic measures. In the
case of unknown causes of neurodegenerative decline, it is imperative that CJD be
included in the differential, and the laboratory testing should be initiated early
in management. There exists a short window of time from symptom onset to death, and
diagnosis early in this window allows the patient to carry out his/her final wishes
and the family time for closure.
Unfortunately, in our patient, as well as the majority of patients who suffer from
CJD, the diagnosis came late in the disease course, after significant neurological
functions had deteriorated and the patient’s quality of life had already been significantly
impacted. Thus, CJD needs to be quickly considered when rapid neurological decline
or NCSE is not suggestive of another entity.
Key Points:
- Creutzfeldt-Jakob disease (CJD), particularly the sporadic subtype, is a devastating
disease with a rapid neurological degeneration pattern.
- In the case of unknown causes of neurodegenerative decline, CJD should be included
in the differential, and the laboratory testing should be initiated early in management.
- A short window of time exists between symptom onset and death, and diagnosis early
in this window allows the patient to carry out his/her final wishes and the family
time for closure.
Acknowledgments
None.
Conflict of Interest Statement & Funding
The authors have no funding, financial relationships, or conflicts of interest to
disclose.
Author Contributions
Conceptualization, Data collection: AJ, JC. Data analysis and interpretation: AJ,
JC, MP, SS. Writing: AJ, JC, DS. Critical revision of the manuscript: AJ, JC, DS,
MP, SS. Approval of the final version: AJ, JC. Contribution of patients or study materials:
MP, SS.
References
1. González-Duarte A, Medina Z, Balaguer RR, Calleja JH. Can prion disease suspicion be supported earlier? Clinical, radiological and laboratory
findings in a series of cases. Prion. 2011 Jul-Sep;5(3):201–7.
2. Head MW, Ironside JW. Review: Creutzfeldt-Jakob disease: prion protein type, disease phenotype and agent
strain. Neuropathol Appl Neurobiol. 2012 Jun;38(4):296–310.
3. Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch Neurol. 2009 Feb;66(2):208–15.
4. Prusiner SB. Shattuck lecture—neurodegenerative disease and prions. N Engl J Med. 2001 May 17;344(20):1516–26.
5. Zerr I, Poser S. Clinical diagnosis and differential diagnosis of CJD and vCJD. With special emphasis on laboratory tests. APMIS. 2002 Jan;110(1):88–98.
6. Espinosa PS, Bensalem-Owen MK, Fee DB. Sporadic Creutzfeldt-Jakob disease presenting as nonconvulsive status epilepticus
case report and review of the literature. Clin Neurol Neurosurg. 2010 Jul;112(6):537–40.
7. Newey CR, Sarwal A, Wisco D, Alam S, Lederman RJ. Variability in diagnosing Creutzfeldt-Jakob disease using standard and proposed diagnostic
criteria. J Neuroimaging. 2013 Jan;23(1):58–63.
8. Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: diagnostic accuracy of CSF 14–3-3 protein in sporadic Creutzfeldt-Jakob
disease: report of the guideline development subcommittee of the American Academy
of Neurology. Neurology. 2012 Oct 2;79(14):1499–506.
9. Bozluolcay M, Elmali AD, Menku SF, Zeydan B, Benbir G, Delil S, et al. Magnetic resonance imaging findings in probable Creutzfeld-Jacob disease: comparison
with electroencephalography and cerebrospinal fluid characteristics. Acta Radiol Short Rep. 2014 Nov 14;3(10):2047981614552218.
Arthur Joseph, 1 Nova Southeastern University College of Osteopathic Medicine, Florida, USA.
Jacob Core, 1 Nova Southeastern University College of Osteopathic Medicine, Florida, USA.
Daniel Solano, 1 Nova Southeastern University College of Osteopathic Medicine, Florida, USA.
Marquand Patton, 2 Palmetto General Hospital, Florida, USA.
Shaun Smart, 2 Palmetto General Hospital, Florida, USA.
About the Author: Arthur Joseph is a student at Nova Southeastern University College of Osteopathic
Medicine.
Correspondence Daniel Solano. Address: Nova Southeastern University College of Osteopathic Medicine,
3301 College Ave, Fort Lauderdale, FL 33314, USA. Email: dsolano088@gmail.com
Cite as: Joseph A, Core J, Solano D, Patton M, Smart S. The importance of rapid consideration of Creutzfeldt-Jakob disease in the differential diagnosis of progressive neurodegenerative disease: a case report. Int J Med Students. 2016 May-Aug;4(2):72-5.
Copyright © 2016 Arthur Joseph, Jacob Core, Daniel Solano, Marquand Patton, Shaun
Smart
International Journal of Medical Students, VOLUME 4, NUMBER 2, August 2016