Review
Mitochondria as a Target for Future Diabetes Treatments
Franziska Thimm1, Marten Szibor2
doi: http://dx.doi.org/10.5195/ijms.2015.115
Volume 3, Number 1: 45-50
Received 24 08 2014:
Accepted 26 01 2015
ABSTRACT
Diabetes mellitus is rapidly becoming the world's most dangerous serial killer. Type
1 diabetes (T1D) is a currently incurable autoimmune disease marked by progressive,
and eventually exhaustive, destruction of the insulin-producing pancreatic beta cells.
Type 2 diabetes (T2D) describes the combination of insulin resistance in peripheral
tissue, insufficient insulin secretion from the pancreatic beta cells, and excessive
glucagon secretion from the pancreatic alpha cells. T1D as well as severe cases of
T2D are treated with insulin replacement, which can merely be considered as life support
for the acute phases of the disease. Islet replacement of insulin-producing pancreatic
beta cells represents a potential treatment method for both insulin-depleted diabetes
(T1D) and insulin-resistant diabetes (T2D) and may shift diabetes management from
life saving measures to a cure. One of the key challenges in islet transplants is
the generation of reactive oxygen species (ROS) and the associated oxidative stress,
which restricts graft longevity. A major leak of ROS takes place during oxidative
phosphorylation at mitochondrial electron transport chain (ETC). Additionally, hyperglycemia-induced
superoxide (O2•-) production has been linked to the development and progression of
diabetic complications, both macrovascular and microvascular. Decreasing ROS in diabetic
patients may prevent the incidence of long term diabetes complications. This review
provides an overview of the role of mitochondria in diabetes, introducing them as
a possible target for future treatment of diabetes.
Keywords:
Reactive Oxygen Species;
Mitochondrial DNA;
Diabetes Mellitus;
Electron Transport;
Oxidative Phosphorylation.
Introduction
The definite etiology of type 1 diabetes (T1D) is still obscure, but considered to
root in a mixture of genetic predisposition and environmental factors, leading to
continued autoimmunity.1 Five percent of diabetics have this type of diabetes.2 In Type 2 diabetes (T2D), peripheral tissue develops a resistance against insulin.3 T2D has been linked to "metabolic syndrome", which is defined by the International
Diabetes Federation (IDF) as central obesity with two of the following: elevated blood
pressure, elevated fasting plasma glucose, high serum triglyce-rides, and low high-density
cholesterol (HDL) levels (Available from: http://www.idf.org/metabolic-syndrome, updated 2014 Oct 23; cited 2015 Jan 21). According to the Centers for Disease Control
and Prevention (CDC), one out of three people will develop T2D in their lifetime.2
The aims of diabetes management are primarily to save life in the short term and secondarily
to prevent the development of diabetic complications in the long term. Both can be
achieved by improving glycemic control, aiming for a glycated hemoglobin (HbA1c) between
6.5%-7.0%.4 For patients with T1D, lifelong insulin replacement therapy marks the therapeutic
objective for attaining glycemic control.5 In T1D, insulin dose varies between 0.4–0.8 UI/kg insulin a day.6 However, insulin administration may induce hypoglycemic episodes that require hospitalization.
The fear of hypoglycemia and the inconvenience of daily insulin injections cause many
patients to neglect proper disease management and experience glycemic lability.7
For the management of T2D, lifestyle adjustments are considered the mainstay. If glycemic
control fails to be obtained by lifestyle changes, treatment may resort to oral hypoglycemic
agents (OHA), including biguanides, sulphonylureas, alpha glucosidase inhibitors,
and thiazolidinediones. In severe cases, insulin injections are required. If indicated,
T2D patients need between 0.2–1.6 UI/kg of insulin a day.5 The current treatment options available for both types of diabetes merely delay the
complications of the disease and do not provide a long-term solution.
Islet replacement of insulin-producing pancreatic beta cells represents a potential
treatment method for both insulin-depleted and insulin-resistant diabetes.8 Following islet transplantation the patients benefit not only from a decrease of
hypoglycemic events, but also spectacular improvement of HbA1c levels and stabilization
of fasting blood glucose, without exposing themselves to the risk of major surgery
as in whole pancreas transplantation.8 A study by Barton and colleagues compared the efficacy of allogenic islet transplantation
in T1D patients in different periods between 1999 and 2010.9 They analyzed 677 T1D patients who had received islet transplants, with the aim to
examine the differences in transplant efficacy between the early (1999-2002), mid
(2003-2006), or recent (2007-2010) transplant era. Three years following the islet
replacement, insulin independence improved from 27% in the early era to 37% and 44%
in the mid and recent eras, respectively, at 36 months after transplantation. Barton
and colleagues suggested that the improvement of graft survival can be linked to more
adequate means for preventing islet rejection.9 One of the key challenges in islet transplantation is the generation of reactive
oxygen species (ROS) and the associated oxidative stress.9 Oxidative stress occurs due to an imbalance between the intracellular free radical
production and the cellular antioxidant defense mechanisms in the transplanted islets,
which can lead to cell death. The cellular antioxidant defense mechanism comprises,
among others, vita-mins and minerals that are hence administered as supplements after
organ transplantation.10 But why not directly target the chief generator of oxidative stress in our cells?
Our mitochondria constitute the major ROS generator, as ROS are released as a byproduct
of oxidative phosphorylation in the electron transport chain (ETC) of the inner mitochondrial
membrane (IMM).
This review provides an overview of the correlation between mitochondria and diabetes,
introducing them as a target for future treatment of diabetes.
Search Strategy and Selection Criteria
A literature search was performed using MEDLINE MeSH terms “reactive oxygen species”,
“alternative oxidase”, “mitochondrial DNA”, “diabetes mellitus”, “respiratory chain”
and “oxidative phosphorylation”. A total of 9807 articles were found. Following filtering
based on selection criteria (publication within last 20 years, English language, and
the condition that at least 4 of the given terms must be in the same article), 7 articles
remained and an additional 13 articles were retrieved from references. This review
follows the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA)
Statement.11
The Powerhouse of the Cell
Mitochondria are of prokaryotic origin and now the powerhouse in eukaryotic cells.
The term was coined by a pioneer in modern cell biology, Philip Siekevitz, and reflects
a critical role of the organelle, which is energy generation by aerobe degradation
of nutrients.10 Mitochondria are enclosed by a double membrane system, the outer mitochondrial membrane
(OMM), and the inner mitochondrial membrane (IMM), each reflecting its function through
its respective structure.12
Metabolic energy is derived through a process known as oxidative phosphorylation.
The proteins required for this process are embedded in the IMM. The surface area that
is required to accommodate the proteins that participate in this process is provided
by the configuration of the IMM into crista (Figure 1).12
Figure 1.
The Mitochondrial Respiratory Chain.
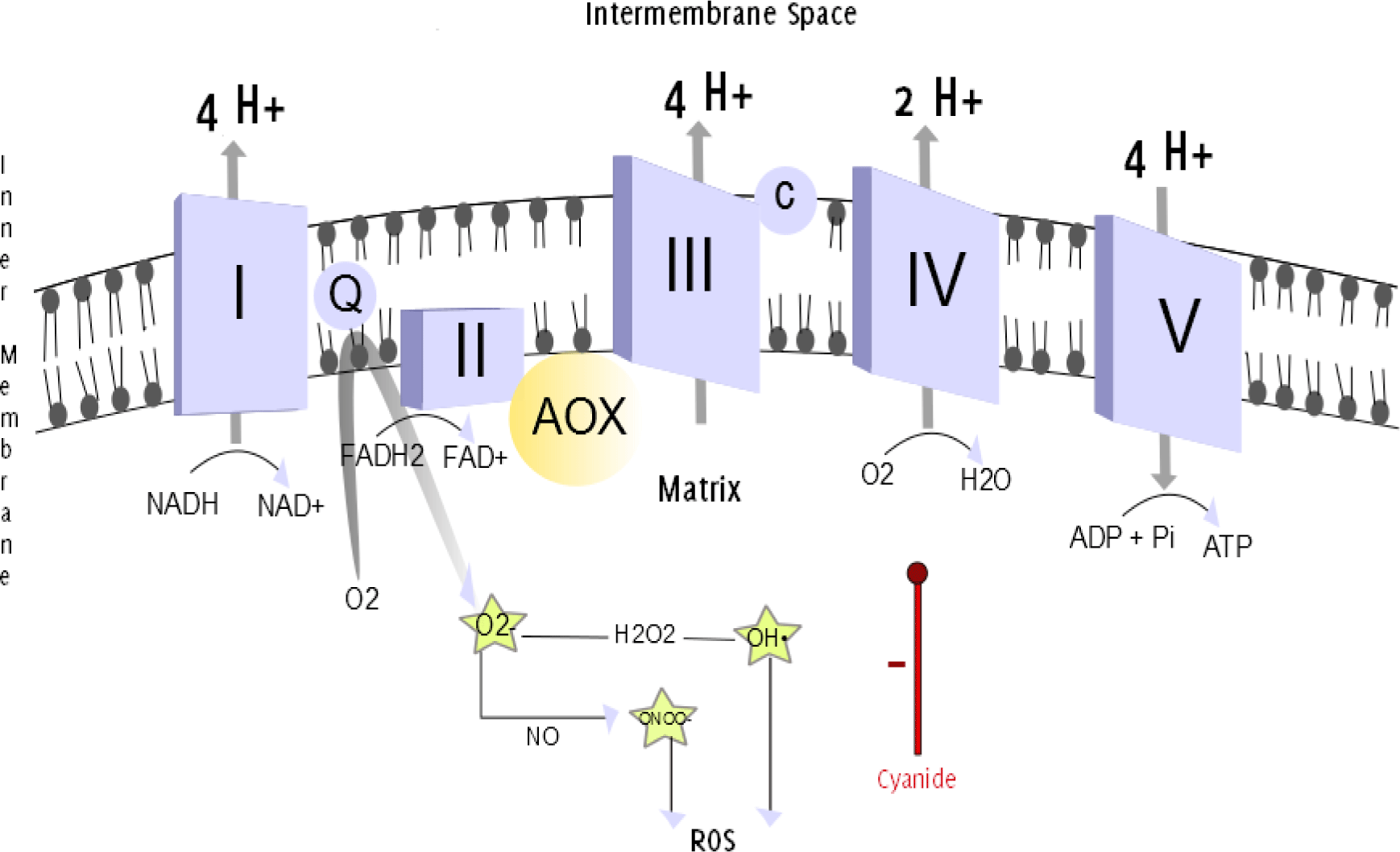
Legend: The mitochondrial respiratory chain. The efflux of protons from the mitochondrial
matrix into the intermembrane space establishes an electrochemical gradient. The back
movement of protons down the electrochemical gradient is utilized by complex V/ATP
Synthase (a proton channel) to phosphorylate ADP, and produce ATP. Oxidation and phosphorylation
are coupled. The uncoupling of the processes can be achieved when uncoupling substances
allow protons to pass through the IMM through an alternative pathway, without passing
through complex V.13–14 This can be realized by the uncoupling protein thermogenin, which is present in brown adipose tissue. This alternative flow results in thermogenesis,
the production of heat, rather than the ATP generation.15 In neonates, thermogenin-containing brown adipose tissue makes up to 5% of the body mass and is located primarily
on the back. This can be explained evolutionarily, as the infant's back was exposed
to the environment during breast feeding and at higher risk of hypothermia.16 AOX provides an alternative pathway for electrons passing through the respiratory
chain, directly converting O2 into H2O, bypassing complexes III and IV. Antimycin and cyanide, inhibitors of complex III and complex IV, respectively, are therefore ineffective
during activation of AOX. Superoxide (O2•-) anions are produced when Coenzyme Q donates
unpaired electrons to molecular oxygen.17 The reaction between O2•- and NO produces peroxynitrite (ONOO•-). Hydrogen peroxide
(H2O2) is converted into a hydroxyl radical (OH•). Both O2•- and ONOO•- are powerful
oxidants.18
Substrate oxidation and adenosine triphosphate (ATP) production [phosphorylation of
adenosine diphosphate (ADP)] are coupled and commonly referred to as "oxidative phosphorylation".12 During oxidation, electrons are transferred from reduced coenzymes (nicotinamide
adenine dinucleotide in its reduced form, NADH, and flavin adenine dinucleotide in
its reduced form, FADH2) via the ETC to molecular oxygen as a final electron acceptor.
The “chemiosmotic hypothesis” of oxidative phosphorylation postulates that during
the electron movement, released energy is used by complexes I, III, and IV to facilitate
the transport of protons from the matrix to the intermembrane space (IMS).19
The proton movement establishes an electrochemical proton gradient (delta Psi, Δψ)
(180-190 mV negative to the cytosol),13 that is subsequently equalized by H+ movement through a gateway of the IMM – “ATP
synthase”. Like in a watermill, the "molecular unit of currency” or ATP is generated
by the movement of protons down their electrochemical gradient through ATP Synthase.14
Oxidative Stress in Diabetes
Oxidative stress makes up two crucial puzzle pieces of diabetes. First, hyperglycemia-induced
long-term complications of diabetes occur due to excessive production of ROS.20 Second, if scientists are successful in decreasing ROS generation after pancreatic
islet replacement, prolonged graft survival could be ensured and diabetes treatment
would shift from being life sustaining to achieving a cure.
Glucose metabolism uses the Krebs cycle to generate NADH and FADH2, which donate electrons
to complex I and complex II, respectively. These electrons are subsequently passed
down the ETC to molecular oxygen as a final electron acceptor, a process which facilitates
proton movement across the IMM. Glucose laden, diabetic cells exhibit a higher rate
of glucose oxidation in the Krebs cycle, triggering more NADH and FADH2 being shoved
into the ETC and ultimately establishing a higher Δψ across the IMM. Eventually the
electrical gradient will reach a threshold, and the electron transfer in complex III
will be inhibited, resulting in the backup of electrons to coenzyme Q. Coenzyme Q
then donates unpaired electrons to molecular oxygen, thereby generating the free radical
O2•-.22
Under the influence of superoxide dismutase (SOD), O2•- can become a weaker ROS, hydrogen
peroxide (H2O2).23 However, when O2•- reacts with nitric oxide (NO•) it may also be converted into the
more active ROS peroxynitrate (ONOO-) (Figure 1).20 Both O2•- and ONOO•- are powerful oxidants.23
Furthermore, hyperglycemia-induced O2•- generation in the mitochondria decreases the
activity of glyceraldehyde-3 phosphate dehydrogenase (GAPDH), an enzyme that serves
as the catalyst for the sixth step of glycolysis and is essential for glucose production
from glycogen breakdown (glycogenolysis).20,24 The inhibition of GAPDH results in the accumulation of glyceraldehyde-3 phosphate,
and the activation of the hyperglycemia pathways described hereafter in detail [protein
kinase C (PKC) pathway; advanced glycation end-products (AGE) pathway].20
The PKC pathway is initiated by the presence of a derivative of glyceraldehyde-3 phosphate,
diacylglycerol (DAG). Excessive intracellular DAG stimulates PKC, a kinase protein
that induces vascular endothelial growth factor (VEGF) expression in non-vascular
cells.25 VEGF is an essential angiogenesis factor and key to neovascularization. The VEGF-mediated
neovascularization of the retina is a process known to play role in diabetic retinopathy.
In diabetic retinopathy, the genesis of friable vessels that frequently rupture results
in hemorrhages that obscure the vision.26 VEGF also increases levels of transforming growth factor beta (TGF-ß), which is profibrinogenic
and accelerates progression to glomerular sclerosis and hypertrophy, contributing
to diabetic nephropathy (Figure 2).27
Figure 2.
Hyperglycemia: The Role of Protein Kinase C Activation in Diabetes Complications.21
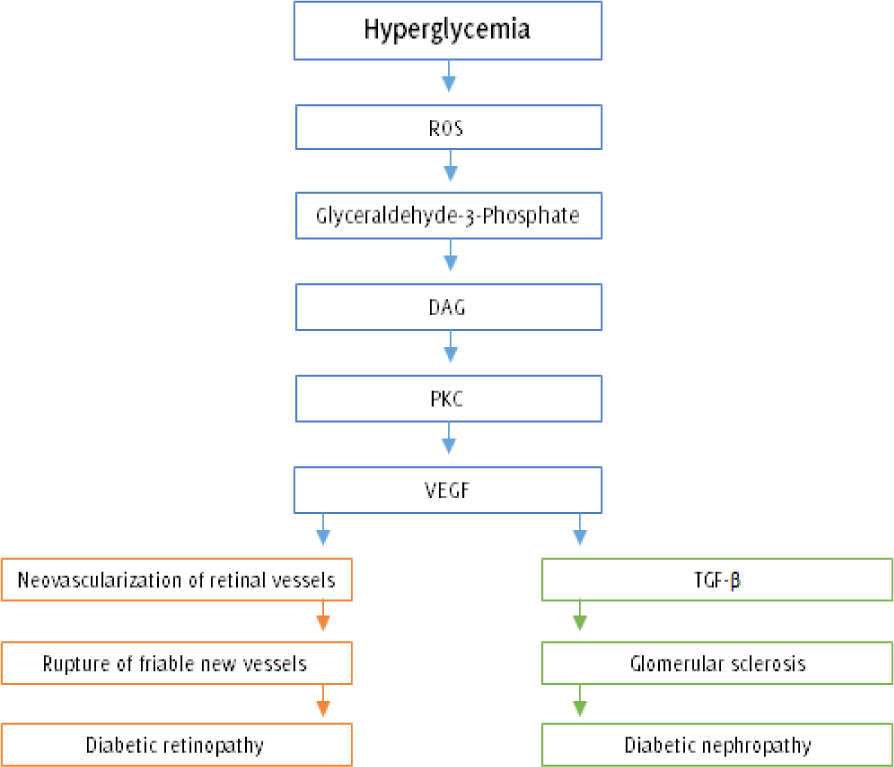
Legend: Hyperglycemia-induced O2•- generation in the mitochondria leads to the accumulation
of glyceraldehyde-3 phosphate and its derivative DAG. Elevated intracellular DAG levels
stimulate the PKC-mediated generation of VEGF, an angiogenesis factor that promotes
neovascularization of retinal vessels. Unfortunately, the newly formed vessels are
highly friable and would often rupture, resulting in hemorrhages that obscure the
vision and destroy the retina (proliferative diabetic retinopathy). On the other hand,
elevated levels of VEGF are responsible for simultaneously rising amounts of TGF-ß
that spurs glomerular proliferation and sclerosis, eventually resulting in diabetic
nephropathy (ROS = Reactive oxygen species; DAG = Diacylglycerol; PKC = Protein kinase
C; VEGF = Vascular endothelial growth factor; TGF-ß = transforming growth factor beta).
Another derivative of glyceraldehyde-3 phosphate, methylglyoxal, activates the AGE
pathway.22 The hyperglycemia-induced AGE pathway generates AGEs by non-enzymatic glycation of
proteins. AGE production is irreversible, and circulating AGEs contribute as a deteriorating
factor to vascular complications of diabetes patients.25
Circulating AGEs are involved in the trapping of albumin, low-density lipoprotein
(LDL), immunoglobulins, and complement components. This procoagulating effect of AGEs
is further increased by its function to deactivate NO•, which leads to the loss of
the vasodilatory effect of NO. Besides, AGEs function as procoagulants themselves
through increasing platelet adhesion and, at the same time, decreasing fibrinolysis.
Also, AGE molecules are capable of binding to RAGE receptors present on both macrophages
and mesangial cells of the kidney. This binding stimulates the release of cytokines
and growth factors, which results in inappropriate cell proliferation, collagen synthesis,
and fibrosis in the glomeruli. Finally, AGEs spur lipid oxidation, which increases
oxidative stress and favors inflammation.28
Many experiments demonstrated that the inhibition of AGEs slow down the developments
of diabetic retinopathy in laboratory rats.29 Hammes and coworkers treated 26-week-old Wistar rats with aminoguanidine, an inhibitor
of AGE formation. The induction of diabetes was achieved with an injection of streptozotocin
in 0.05 M sodium citrate. The administration of aminoguanidine was initiated 2 weeks
later. Advanced glycosylation-specific fluorescence enabled the scientists to visualize
the amount of accumulated AGEs in the animals' retinal vessels in the 26th and 75th
week. While no morphological changes and 170±15 fluorescence absorbance units were
observed in the healthy animals after 75 weeks, the diabetic rats had a high degree
of neovascularization with friable vessels and 440±20 fluorescence absorbance units.
The retinal vessels of those diabetic animals that had received aminoguanidine injections
were less affected by these morphological changes, and glycosylation product-specific
fluorescence measured was only 220±13.30 Although aminoguanidine has been under development as a drug by the pharmaceutical
company Alteon, clinical trials have been abandoned since 1998.31
The inhibition of GAPDH also results in the accumulation of the first and second glycolytic
metabolites, glucose and fructose 6-phosphate. Glucose is reduced by aldose reductase
into sorbitol, which is further oxidized by the enzyme sorbitol dehydrogenase into
fructose. This so-called polyol pathway leads to a decrease of reduced NADPH.26 NADPH is a crucial cofactor in redox reactions throughout the body, including the
synthesis of myoinositol in the kidneys (Figure 3). Myoinositol deficiency has been shown to be present in laboratory animals with
induced diabetes as well as the sciatic nerve from deceased diabetic patients.26
Figure 3.
The Polyol Pathway in Hyperglycemia.35
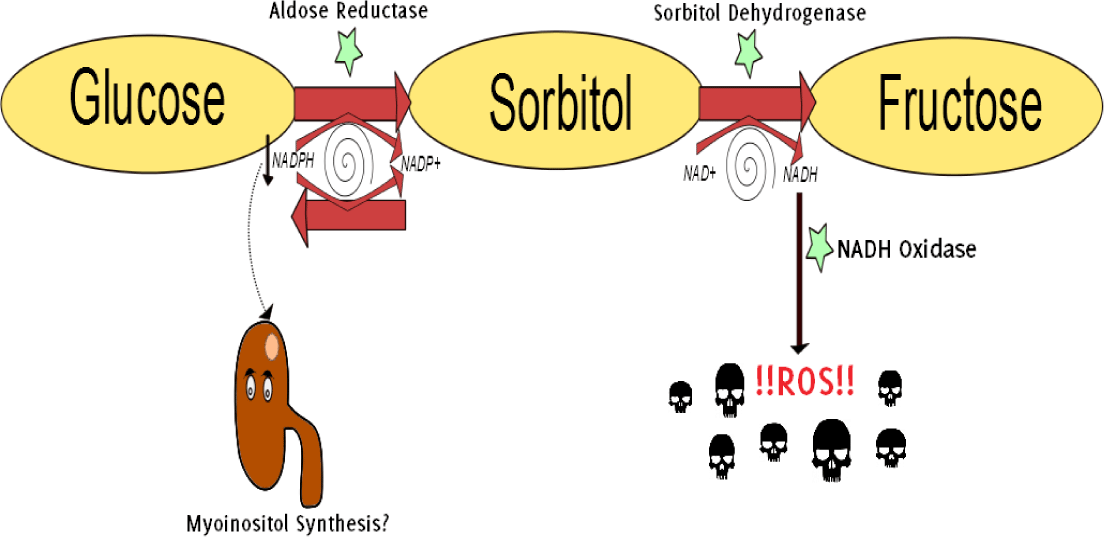
Legend: Hyperglycemia stimulates the aldose reductase-mediated reduction of glucose into
sorbitol (a polyol, hence polyol pathway). Aldose reductase utilizes cofactor NADPH,
which results in a depletion of the antioxidant glutathione (GSH) and increases intracellular
oxidative stress.31 Besides, NADPH is a crucial cofactor in redox reactions throughout the body, including
the synthesis of myoinositol in the kidneys. Myoinositol deficiency has been identified
in diabetic patients.36 Sorbitol is further oxidized to fructose. The generated NADH is subsequently converted
by NADH oxidase into ROS and, therefore, acts as a booster for oxidative stress on
the cell.37
Myoinositol is particularly important for the normal function of nerves. Already in
1987 it has been suggested by Salway and colleagues that myoinositol may prove valuable
in preventing or delaying diabetic neuropathy. Neuropathy is marked by a slowed conduction
velocity in peripheral and autonomic nerve fibers. Salway and his team, who had administered
500 mg myoinositol twice a day to seven different diabetes patients over a time span
of two weeks, had observed an increased amplitude of the action potential of three
different nerves (two in the lower extremity, one in the upper extremity). They suggested
the possible value of myo-inositol in diabetes treatment in the future.30
In excess, ROS can activate several stress-sensitive intracellular signaling pathways
(NF-kB, p38 MAPK, JNK/SAPK, and hexosamine) that induce gene expression. The products
of these genes are involved not only in the development of diabetes complications,
but also the development of insulin resistance.32,33 For example, hyperglycemia-induced overproduction of O2•-diverts fructose-6-phosphate
to the hexosamine pathway. The end product of the hexosamine pathway, UDP-N-acetylglucosamine
is capable of glycating intracellular proteins, including transcription factors that
alter gene expression (Available from: http://www.thepharmaletter.com/article/alteon-may-drop-pimagedine-in-niddm, updated 2014 Dec 21, cited 2015 Jan 21). This suggests that antioxidants could play
a role in delaying and/or preventing the development of diabetes complications and
insulin resistance, thereby immensely altering the pathophysiology of T2D.
Alternative oxidase (AOX)
Alternative oxidase (AOX) is an integral mitochondrial membrane enzyme which is mainly
found in sessile organisms. It is capable of limiting the overproduction of mitochondrial
ROS. However, this alternative pathway bypasses several proton-pumping steps, decreasing
the pH and electrical gradients, thereby reducing the ATP generation. During the course
of evolution, AOX has been lost from fast-moving organisms (including humans), maybe
due to the slight reduction in ATP production.34
By bypassing the respiratory chain, AOX confers resistance to cyanide and other inhibitors
of the respiratory chain. Sessile animals, deep-sea organisms which are exposed to
a hostile environment, thereby benefit from AOX and are still endowed with it.34
AOX provides a bypath of the ETC, thereby decreasing the O2•- generation and suppressing
the infliction of oxidative damage on the cell. In the lab, AOX has been safely expressed
in flies and human cells without eliciting any unwanted physiological side effects.34
In 2013, C. intestinalis' (a sessile sea squirt) AOX gene was successfully expressed in mouse embryos with
the help of germ line lentiviral transduction and subsequently passed down to the
next generation.38 This so-called MitAOX mouse could be crossed with selective lines of diabetic mice,
for example the non-obese diabetic (NOD) mice, an animal model for T1D. The resultant
diabetic mice should have AOX incorporated in their genome. This AOX incorporation
could be helpful to investigate hyperglycemia-induced overproduction of O2•- and its
role in the development of diabetic complications.
Additionally, it has been suggested that AOX mitochondrial targeting sequence could
be delivered with the aid of a viral vector, especially since recent data clearly
points to the longevity and safety of viral vectors.34 This injectable AOX has the potential to allow for therapeutic application in many
disorders with marked overproduction of O2•-.39
Oxidative stress is influencing the allograft outcome during the peritransplantation
period of kidney transplant.38 In a study that Morales-Indiano and colleagues conducted on 131 patients with end
stage renal disease (ESRD), both diabetic and non-diabetic patients had similar oxidative
stress levels before kidney transplantation. However, measured oxidative stress was
significantly higher in the diabetic patients after the intervention. In order to
effectively measure oxidative stress, Morales-Indiano and colleagues determined and
measured oxidative stress markers both prior to and at 120 days after grafting. The
markers used to measure the generation of oxidative stress included anti-oxLDL antibodies
(oxLDLab) and oxidized LDL. The poorer allograft function in diabetics was attributed
to elevated HbA1C, which promotes oxidative stress.17
In future, AOX injections may be administered to decrease hyperglycemia-induced oxidative
stress in diabetic patients after transplant, including pancreatic islet transplants.
Acknowledgments
University of Helsinki, Mitochondrial Gene Expression and Disease group by Howard
Jacobs.
Conflict of Interest Statement & Funding
The author has no funding, financial relationships or conflicts of interest to disclose.
Author Contributions
Conception and design the work/idea, Collect data/obtaining results, Analysis and
interpretation of data, Write the manuscript, Critical revision of the manuscript,
Approval of the final version: FT, MS.
References
1. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2004 Jan;27 suppl 1:S5–S10.
2. Centers for Disease Control and Prevention. National Diabetes Statistics Report 2014: Estimates of diabetes and its burden in
the United States. Atlanta, GA: U.S. Department of Health and Human Services. Report number: 1, 2014.
3. Kadowaki T. Insights into insulin resistance and type 2 diabetes from knockout mouse models. J Clin Invest. 2000 Aug;106(4):459–65.
4. Bailey CJ, Blonde L, Del Prato S, Leiter LA, Nesto R; Global Partnership for Effective Diabetes Management. What are the practical implications for treating diabetes in light of recent evidence?
Updated recommendations from the Global Partnership for Effective Diabetes Management. Diab Vasc Dis Res. 2009 Oct;6(4):283–7.
5. Bastaki S. Diabetes mellitus and its treatment. Int J Diabetes & Metabolism. 2005;13(3):111–34.
6. Jakhmola V, Tangri P. Diabetes Mellitus a silent killer: Role of DPP 4 inhibitors in treatment. JPSBR. 2012 Mar-Apr; 2(2):49–53.
7. Dardano A, Bianchi C, Del Prato S, Miccoli R. Insulin degludec/insulin as-part combination for the treatment of type 1 and type
2 diabetes. Vasc Health Risk Manag. 2014 Aug;10(1):465–75.
8. Chhabra P, Brayman KL. Current status of immunomodulatory and cellular therapies in preclinical and clinical
islet transplantation. J Transplant. 2011, 2011:637692.
9. Barton FB, Rickels MR, Alejandro R, Hering BJ, Wease S, Naziruddin B et al.. Improvement in outcomes of clinical islet transplantation: 1999–2010. Diabetes Care. 2012 Jul;35(7):1436–45.
10. Ramkumar KM, Sekar TV, Bhakkiyalakshmi E, Foygel K, Rajaguru P, Berger F et al.. The impact of oxidative stress on islet transplantation and monitoring the graft survival
by non-invasive imaging. Curr Med Chem. 2013;20(9):1127–46.
11. Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Loannidis JP et al.. The PRISMA statement for reporting systematic reviews and meta-analyses of studies
that evaluate health care interventions: explanation and elaboration. PLoS Med. 2009Jul21;6(7):e1000100.
12. Papa S, Martino PL, Capitanio G, Gaballo A, De Rasmo D. The oxidative phosphorylation system in mammalian mitochondria. In: Scatena R, Bottoni P, Giardina B, editors. Advances in mitochondrial Medicine. 1st ed. Dordrecht: Springer; 2012. p. 3–38.
13. Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967Jan14;213(5072):137–9.
14. Alberts B, Johnson A, Lewis J, Raff M, Roberts K. Energy conversion: Mitochondria and chloroplasts. In: Alberts B, Johnson A, Lewis J, Raff M, Roberts K, et al., editors. Molecular biology of the cell. 5th ed. New York: Garland Science; 2007. p. 813–78.
15. Morales-Indiano C, Lauzurica R, Pastor MC, Bayés B, Sancho A, Troya M et al.. Greater posttransplant inflammation and oxidation are associated with worsening kidney
function in patients with pretransplant diabetes mellitus. Transplant Proc. 2009 Jul-Aug; 41(6):2126–8.
16. Ricquier D, Bouillaud F. The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J. 2000Jan15;345 Pt 2:161–79.
17. Nafar M, Sahraei Z, Salamzadeh J, Samavat S, Vaziri ND. Oxidative stress in kidney transplantation: causes, consequences, and potential treatment. Iran J Kidney Dis. 2011 Nov;5(6):357–72.
18. Carter BW, Schucany WG. Brown adipose tissue in a newborn. Proc (Bayl Univ Med Cent). 2008 Jul; 21(3):328–30.
19. Cali T, Ottolini D, Brini M. Mitochondrial Ca2+ as a key regulator of mitochondrial activities. In: Scatena R, Bottoni P, Giardina B, editors. Advances in mitochondrial medicine. 1st ed. Dordrecht: Springer. 2012. p. 53–73.
20. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005 Jun;54(6):1615–25.
21. Chung SS, Ho EC, Lam KS, Chung SK. Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol. 2003 Aug;14(suppl 3):S233–6.
22. Knowles JR. Enzyme-catalyzed phosphoryl transfer reactions. Annu Rev Biochem. 1980 Jul; 49(1):877–919.
23. Shen GX. Mitochondrial dysfunction, oxidative stress and diabetic cardiovascular disorders. Cardiovasc Hematol Disord Drug Targets. 2012 Dec;12(2):106–12.
24. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003Oct15;552(Pt 2):335–44.
25. Xu H, Czerwinski P, Hortmann M, Sohn HY, Förstermann U, Li H. Protein kinase C alpha promotes angiogenic activity of human endothelial cells via
induction of vascular endothelial growth factor. Cardiovasc Res. 2008May1;78(2):349–55.
26. Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M. Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci U S A. 1991 Dec 15;88(24):11555–8.
27. Berg JM, Tymoczko JL, Stryer L. Glycolysis and gluconeogenesis. In: Ahr K, Baker A, Tymoczko N, Goldman D, Moscatelli B, et al., editors. Biochemistry. 6th ed. New York: W. H. Freeman. 2006. p. 433–74.
28. Thallas-Bonke V, Cooper ME. Tandem inhibition of PKC in Diαβetic nephropathy: it takes two to tango? Diabetes. 2013 Apr;62(4):1010–1.
29. Yan SF, D'Agati V, Schmidt AM, Ramasamy R. Receptor for advanced glycation endproducts (RAGE): a formidable force in the pathogenesis
of the cardiovascular complications of diabetes & aging. Curr Mol Med. 2007 Dec;7(8): 699–710.
30. Holub BJ. Metabolism and function of myo-inositol and inositol phospholipids. Annu Rev Nutr. 1986 Jul;6(1):563–97.
31. Maitra A. Endocrine system. In: Kumar V, Abbas AK, Aster JC, editors. Robbins basic pathology. 9th ed. Philadelphia: Elsevier Saunders. 2013. p. 715–64.
32. Salway JG, Whitehead L, Finnegan JA, Karunanayaka A, Barnett D, Payne RB. Effect of myo-inositol on peripheral-nerve function in diabetes. Lancet. 1978 Dec 16;2(8103):1282–4.
33. Newsholme P, Haber EP, Hirabara SM, Rebelato EL, Procopio J, Morgan D et al.. Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial
ROS production and activity. J Physiol. 2007Aug15;583(Pt 1):9–24.
34. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance
and beta-cell dys-function? Diabetes. 2003 Jan;52(1):1–8.
35. Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010Apr30;106(8):1319–31.
36. Squadrito GL, Pryor WA. Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon
dioxide. Free Radic Biol Med. 1998 Sep;25(4-5):392–403.
37. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001 Dec 13;414(6865):813–20.
38. El-Khoury R, Kemppainen KK, Dufour E, Szibor M, Jacobs HT, Rustin P. Engineering the alternative oxidase gene to better understand and counteract mitochondrial
defects: state of the art and perspectives. Br J Pharmacol. 2014 Apr;171(8):2243–9.
39. Bouaita A, Augustin S, Lechauve C, Cwerman-Thibault H, Benit P, Simonutti M et al.. Downregulation of apoptosis-inducing factor in Harlequin mice induces progressive
and severe optic atrophy which is durably prevented by AAV2-AIF1 gene therapy. Brain. 2012 Jan;135(Pt 1):35–52.
Franziska Thimm, 1 Medical Student, University of Latvia, Latvia.
Marten Szibor, 2 Mitochondrial Gene Expression and Disease Group, University of Helsinki, Finland.
About the Author: Franziska Thimm is currently a 4th year medical student at the University of Latvia,
Riga, Latvia of a 6 year program. She is also a former Editor-in-Chief of the European
Medical Student Association's (EMSA) official magazine EuroMeds.
Correspondence Franziska Thimm, Address: Raiņa bulvāris 19, Rīga, LV-1586, Latvia. Email: franziska.thimm@gmail.com
Cite as: Thimm F, Szibor M. Mitochondria as a Target for Future Diabetes Treatments. Int J Med Students. 2014 Nov-2015 Mar;3(1):45-50.
Copyright © 2015 Franziska Thimm, Marten Szibor
International Journal of Medical Students, VOLUME 3, NUMBER 1, March 2015